From: Robert Johnson (robertjo_at_physics.upenn.edu)
Date: Fri Jul 13 2012 - 13:35:46 CDT
Hello Everyone,
I have some questions about the use of the extended Lagrangian feature in
NAMD. Below are links to 3 images of ABF calculations using the RMSD to a
DNA reference structure as the collective variable. I apologize for the
lack of axis labels, but the vertical axis in each plot is the PMF and the
horizontal axis is the RMSD to the reference structure in angstroms.
http://www.physics.upenn.edu/~robertjo/images/GCGC_ExtLagrangeOn.jpg
http://www.physics.upenn.edu/~robertjo/images/GCGC_ExtLagrangeOff.jpg
http://www.physics.upenn.edu/~robertjo/images/GTAC_ExtLagrangeOn.jpg
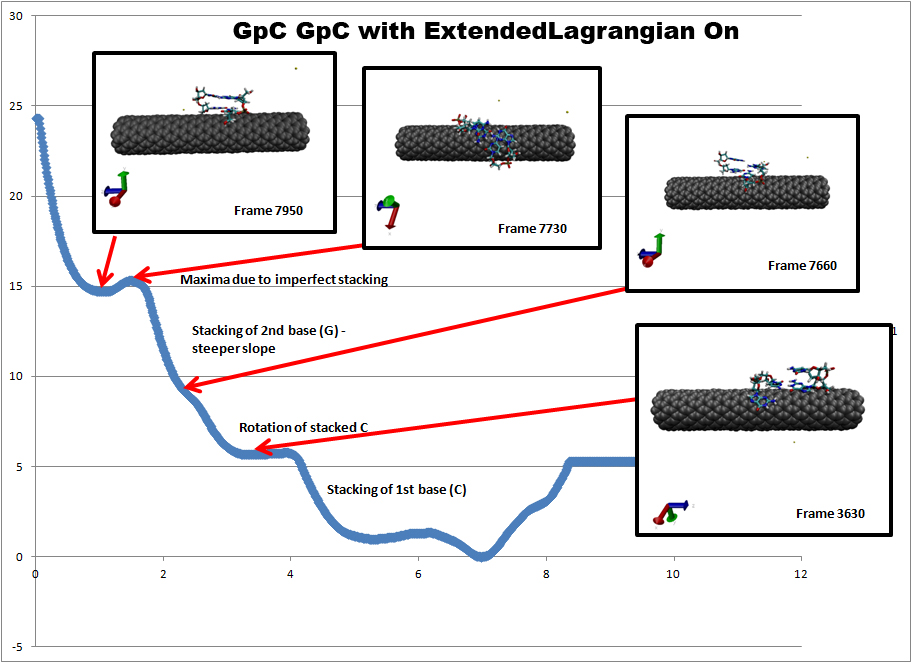
For a system involving two self-complementary DNA strands (in this case GpC
GpC), we get a PMF that contains several energy minima that are associated
with specific physical changes in our system. This can be seen in the first
image (http://www.physics.upenn.edu/~robertjo/images/GCGC_ExtLagrangeOn.jpg).
The main feature here is that the system exhibits two well-defined energy
minima: one for the unhybridized state where the DNA is fully adsorbed to
the carbon nanotube and one with the DNA fully hybridized.
This PMF was obtained using the following parameters:
extendedLagrangian on
extendedTimeConstant 200
extendedFluctuation 0.1
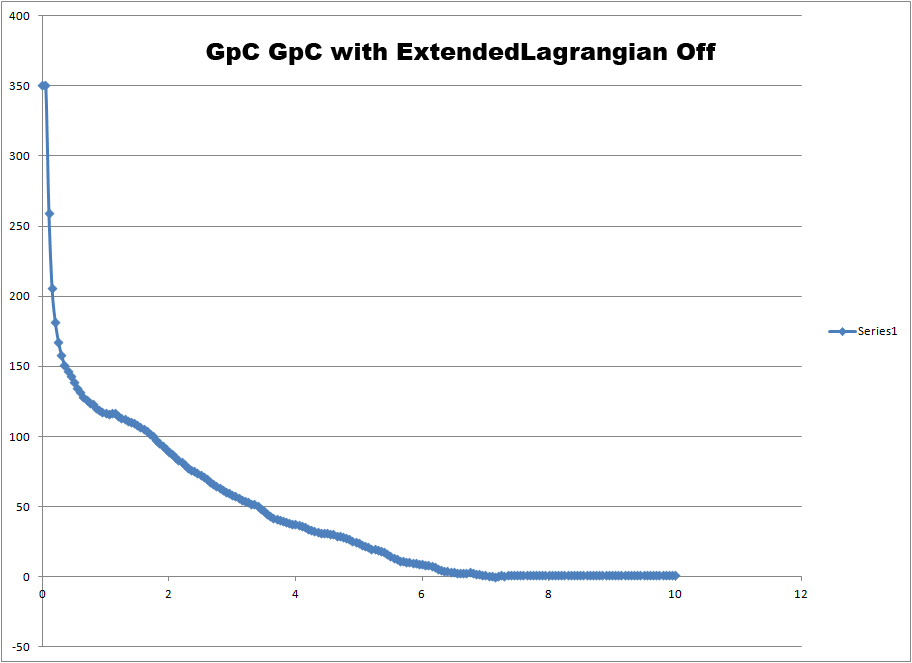
We are using NAMD 2.8 and the parameter extendedLangevinDamping is not
recognized in the version we are using. It seems that using
extendedLagrangian is important for this system. With this feature turned
off we obtain a PMF with very few features. This can be seen in the second
image (http://www.physics.upenn.edu/~robertjo/images/GCGC_ExtLagrangeOff.jpg).
Notice that the second energy minimum is no longer present.
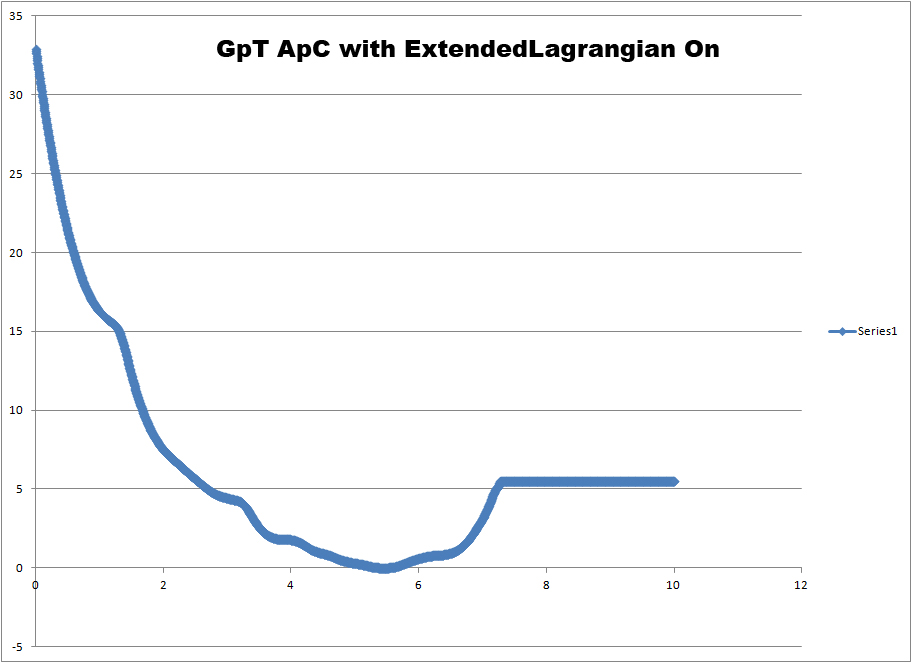
I'm interested to know what this extended Lagrangian is doing and why it is
important. We have tried running the exact same simulation with
extendedLagrangian on for a different system where the two sequences are
GpT and CpA. When we run this simulation we get a PMF that is somewhat
similar to the one for the GpC GpC system with extendedLagrangian off.
Namely, there is no longer an energy minima associated with the final DNA
hybridized state. This can be seen in the third image (
http://www.physics.upenn.edu/~robertjo/images/GTAC_ExtLagrangeOn.jpg).
Based on my intuition of the system, there should be a local minima with
GpT ApC. However, as the PMF shows, the free energy just keeps on going up
and up as the DNA is forced to hybridize.
Does anyone have ideas about why this could be? Does it have to do with the
choice of parameters for this extended Lagrangian method? Any insights or
suggestions would be great!
Thanks,
Bob
On Thu, Jul 5, 2012 at 6:24 AM, Jérôme Hénin <jhenin_at_ifr88.cnrs-mrs.fr>wrote:
> Hi Bob,
>
> In an ideal world, you'd use a multiple-walker ABF formalism to
> explore all these pathways at once, in parallel. It is documented in
> principle (http://pubs.acs.org/doi/abs/10.1021/ct900524t) but there is
> no implementation that is usable enough for your purpose.
>
> Averaging data after the fact seems very legitimate to me. Just don't
> average the PMFs, instead, compute a weighted average of the
> gradients, and integrate the result. You can let the ABF code do that
> for you: run a 0-step ABF simulation where you provide all of the
> previous ABF outputs through the inputPrefix keyword. This will read
> and combine the data properly and give you a single output.
>
> Cheers,
> Jerome
>
>
> On 3 July 2012 20:47, Robert Johnson <robertjo_at_physics.upenn.edu> wrote:
> > Hello Everyone,
> > Are there any best practices for obtaining the average PMF from multiple
> > runs? I am now using the RMSD to a reference structure as my collective
> > variable. This has greatly improved the ability of my system to reach the
> > desired endpoint. However, because DNA is very flexible there are many
> > different pathways that can be taken to reach the final endpoint. As a
> > result, each run results in a slightly different PMF with a different
> value
> > of the free energy difference between my initial and final states. I have
> > played around with the fullSamples and width parameters: right now I'm
> using
> > fullSamples 1000 and width 0.005A. I think converging on a single PMF is
> > just not possible in a single run with my system because it is so
> flexible.
> > My current plan is to run the calculation several times to get an idea
> about
> > the ensemble of PMFs that characterize the system and then just average
> them
> > to get "the" PMF for the process. Does this sound like a good approach?
> Are
> > there any other things to consider?
> > Thanks,
> > Bob
> >
> >
> > On Thu, Jun 21, 2012 at 10:47 AM, Jérôme Hénin <jhenin_at_ifr88.cnrs-mrs.fr
> >
> > wrote:
> >>
> >> Hi Bob,
> >>
> >> One caveat with the RMSD variable is to use small bins (smaller than
> >> for a distance, typically). 0.05 A has worked for me in the past, but
> >> in principle it depends on the ruggedness of the PMF.
> >>
> >> Cheers,
> >> Jerome
> >>
> >>
> >> On 20 June 2012 18:30, Robert Johnson <robertjo_at_physics.upenn.edu>
> wrote:
> >> > Hi Jerome,
> >> > Your idea of using the RMSD sounds like a good one to me. We don't
> >> > expect to
> >> > get a rigorous result for the PMF - we are more interested in
> >> > qualitative
> >> > results. I've never used the RMSD as a collective variable. I see
> there
> >> > is
> >> > documentation on how to do this here:
> >> >
> >> >
> http://www.ks.uiuc.edu/Research/namd/2.9/ug/node55.html#SECTION0001322150000000000000
> >> >
> >> > I also saw that there was some previous discussion on how to do this
> on
> >> > the
> >> > mailing list:
> >> > http://www.ks.uiuc.edu/Research/namd/mailing_list/namd-l/12123.html
> >> >
> >> > The user mentions that he is following the tutorial for ubiquitin. I
> >> > found a
> >> > tutorial here:
> http://www.ks.uiuc.edu/Training/CaseStudies/pdfs/ubq.pdf
> >> > However, it seems that the only colvar that is used is the end-to-end
> >> > distance and not the RMSD. Is there another tutorial available?
> >> >
> >> > In the meantime we will try to follow the instructions in the user
> guide
> >> > and
> >> > perhaps we can get it to work on the first try. I'm just wondering if
> >> > there
> >> > are any other caveats that I need to worry about when using this type
> of
> >> > colvar.
> >> > Thanks,
> >> > Bob
> >> >
> >> >
> >> > On Wed, Jun 20, 2012 at 7:25 AM, Jérôme Hénin <
> jhenin_at_ifr88.cnrs-mrs.fr>
> >> > wrote:
> >> >>
> >> >> Hi Bob,
> >> >>
> >> >> As you've noticed, the coordinate you used so far gives ambiguous
> >> >> results because your system has a lot of flexibility, and will visit
> >> >> basins that are not of interest to you. Now there are two kinds of
> >> >> approaches to this problem:
> >> >>
> >> >> 1) add restraints that forbid visiting the unwanted states, but this
> >> >> changes the meaning of the PMF you are calculating
> >> >> 2) change your set of coordinates to describe the space of interest
> >> >> more explicitly, and explore precisely that
> >> >>
> >> >> In many cases where you want mostly qualitative information on a
> >> >> precise process, the first choice is the best one. Trying to extract
> a
> >> >> PMF that is quantitative and meaningful and can yield real free
> energy
> >> >> differences can be very demanding.
> >> >>
> >> >> Now about finding coordinates that describe the process: one simple
> >> >> coordinate that would discriminate between the states that you
> mention
> >> >> is the RMSD of the whole dimer with respect to the hybridized state--f46d042c647736d01f04c4ba59e4--
This archive was generated by hypermail 2.1.6 : Tue Dec 31 2013 - 23:22:15 CST
{kind=link}
{kind=link}
{kind=link}