From: Francesco Pietra (chiendarret_at_gmail.com)
Date: Mon Mar 07 2011 - 10:04:37 CST
Hello Peter:
Thanks for answering. Please, see below.
On Mon, Mar 7, 2011 at 4:19 PM, Peter Freddolino <petefred_at_ks.uiuc.edu> wrote:
> Hi Francesco,
>
> On 03/07/2011 01:12 AM, Francesco Pietra wrote:
>>
>> this led to the added proton being in the unfavorable position that I
>> described, although the psf/pdb could be minimized and heated.
>
> Could you elaborate on how unfavorable this position seems? What's wrong
> with it? Unless you have something truly pathological, a minimization
> should give you an appropriate orientation.
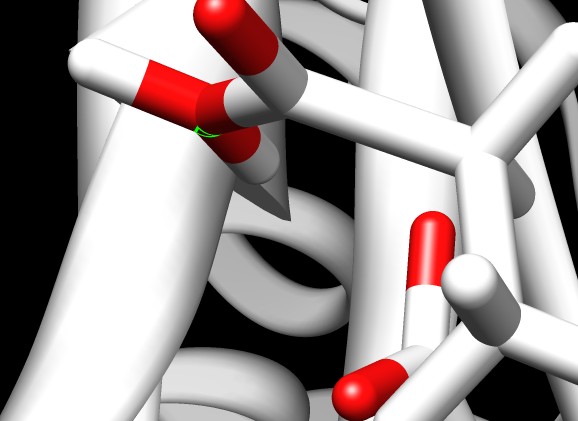
Attached please see a picture of a GLU-ASP couple. At OE2 (highlighted
in cyan) of GLU I have attached a second proton (syn to ASP), at 180
deg from the original one (anti to ASP). That should illustrate that
the original placement of the proton is unfavorable for H-bonding with
ASP. Minimization should not (I guess) place the proton syn to ASP
because the original anti position has no unfavorable interactions.
> psfgen has no knowledge of
> the force field, so it won't even try to place the hydrogen in a way
> that forms hydrogen bonds.
In fact, while I am examining in detail all H-bonds, I noticed that -
contrary to what I said before - not all protons have been placed
anti. It is fifty:fifty anti/syn, as for random placements.
>>
>> Previously, I also filled in "Autopsf - Add Patch", in addition to the above,
>>
>> Segment 2 (opt) ... Residue 2:....
>>
>> for the intended acceptor of Segment 1, Residue 1
>>
>> at no avail as far as the H-bond is concerned. This latter action in
>> an imitation of the DISU patch, where both S atoms of the S-S bond are
>> set in.
>
> Specifying a second residue does no good whatsoever... the topology
> entry that you're using only acts on one residue.
It was a sort of desperate attempt from me, as I could not fully
understand the meaning of the option.
I am now trying again to set up in the pdb file from autopsf what I
believe is the correct stereochemistry, to run autopsf again. Maybe I
did a mistake in a previous similar attempt. However, I would be not
too much surprised that the matter can be tackled differently. These
are problems that people should have encountered each other day.
Thanks a lot
francesco
>
>> ************
>>
>> What I hope is that there is a mistake in my procedure, and be
>> corrected about. Otherwise suggestions how to set correctly H-bonds in
>> VMD/NAMD along a different route. As I said, I came to NAMD with
>> correct PDB files - from REDUCE or other - as far as H-bonds are
>> concerned. However, files strictly respecting PDB rules and, in
>> addition, also with some non CHARMM naming, which I tried to correct.
>> I was unable to arrive at workable psf/pdb along this route. Finally,
>> I could also correct the autopsf.pdb by repositioning the proton in
>> between GLU and the acceptor, be that the conjugate base or Cl-, with
>> a graphic package. However, this also failed - in my hands - to arrive
>> at workable psf/pdb.
>
> You would again need to give more details on what you tried to do and
> what errors occurred in order to get help.
>
> Best,
> Peter
>
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:19:54 CST