From: Gadi Oron (gadi_at_proteologics.com)
Date: Tue Dec 16 2003 - 09:33:25 CST
Hello,
On Fri, 2003-12-12 at 11:16, Alexandre Vakhrouchev wrote:
Thank you for your reply.
>
> First of all look at the output of your psfgen script (it is attached
> as 'pfs.log').
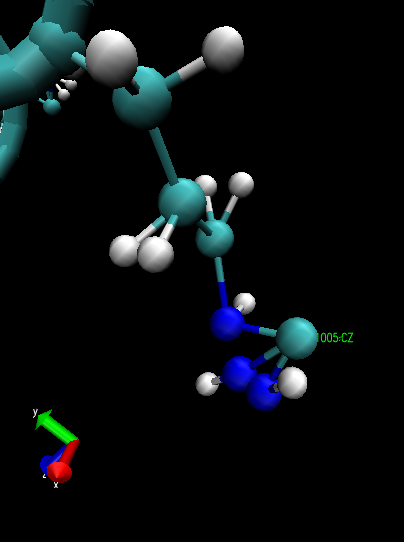
I have noticed that some coordinates are guessed, but I don't think that
this is the reason for the crash. You are right though that the the
crash is due to close atoms, I have written a script that finds atoms
too close to each other, and found that at the end of the minimization
some atoms are literally on top of each other. The problem is that this
was not the case before the minimization! ARG residues are especially
bizarre. Their atoms simple collapse during minimization to distances
less than half an A(!!). I have attached a snapshot to illustrate it.
I do believe that this in NOT due to the patches that I apply since
before minimization everything is fine. I have also isolated a single
chain that is not patched or modified, and it behaves the same (chain
C).
The only way I could get the dynamics running is to specify
"exclude none", but in this case the protein swells into unrealistic
dimensions (H bonds become 2A long!)...
Any ideas?
> I solved this error by providing coors for all atoms,
> depending on Rmin/2 values (from Van-der-Waals force field
> parametres), but i had to
> write additional programm.
I would be glad to have this prog... even though I doubt it would be of
great help...
Thank you for your help.
-- ================= Gadi ORON, PhD -o- Proteologics ====================== =========== gadi/at/proteologics.com =========== +972 8 9475666 =========== Also, the Scots are said to have invented golf. Then they had to invent Scotch whiskey to take away the pain and frustration.
This archive was generated by hypermail 2.1.6 : Wed Feb 29 2012 - 15:37:14 CST