



Next: User Defined Forces
Up: Standard Minimization and Dynamics
Previous: Temperature Control and Equilibration
Contents
Index
Subsections
Constant pressure simulation (and pressure calculation) require periodic
boundary conditions. Pressure is controlled by dynamically adjusting
the size of the unit cell and rescaling all atomic coordinates (other than
those of fixed atoms) during the simulation.
Pressure values in NAMD output are in bar.
PRESSURE is the pressure calculated based on individual atoms, while
GPRESSURE incorporates hydrogen atoms into the heavier atoms to which
they are bonded, producing smaller fluctuations.
The TEMPAVG, PRESSAVG, and GPRESSAVG are the average of temperature and
pressure values since the previous ENERGY output; for the first step
in the simulation they will be identical to TEMP, PRESSURE, and GPRESSURE.
The phenomenological pressure of bulk matter reflects averaging in both
space and time of the sum of a large positive term (the kinetic pressure,
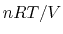 ), and a large cancelling negative term (the static pressure).
The instantaneous pressure of a simulation cell as simulated by NAMD
will have mean square fluctuations (according to David Case quoting
Section 114 of Statistical Physics by Landau and Lifshitz)
of
), and a large cancelling negative term (the static pressure).
The instantaneous pressure of a simulation cell as simulated by NAMD
will have mean square fluctuations (according to David Case quoting
Section 114 of Statistical Physics by Landau and Lifshitz)
of
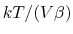 , where
, where 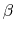 is the compressibility, which is
RMS of roughly 100 bar for a 10,000 atom biomolecular system.
Much larger fluctuations are regularly observed in practice.
is the compressibility, which is
RMS of roughly 100 bar for a 10,000 atom biomolecular system.
Much larger fluctuations are regularly observed in practice.
The instantaneous pressure for a biomolecular system is well defined for
``internal'' forces that are based on particular periodic images of the
interacting atoms, conserve momentum, and are translationally invariant.
When dealing with externally applied forces such as harmonic constraints,
fixed atoms, and various steering forces, NAMD bases its pressure calculation
on the relative positions of the affected atoms in the input coordinates
and assumes that the net force will average to zero over time. For time
periods during with the net force is non-zero, the calculated pressure
fluctuations will include a term proportional to the distance to the
affected from the user-defined cell origin.
A good way to observe these effects and to confirm that pressure for
external forces is handled reasonably is to run a constant volume cutoff
simulation in a cell that is larger than the molecular system by at least
the cutoff distance; the pressure for this isolated system should average
to zero over time.
Because NAMD's impluse-basd multiple timestepping system alters the
balance between bonded and non-bonded forces from every timestep to an
average balance over two steps, the calculated pressure on even and odd
steps will be different. The PRESSAVG and GPRESSAVG fields provide the
average over the non-printed intermediate steps. If you print energies on
every timestep you will see the effect clearly in the PRESSURE field.
The following options affect all pressure control methods.
NAMD provides constant pressure simulation using Berendsen's method.
The following parameters are used to define the algorithm.
- BerendsenPressure
 use Berendsen pressure bath coupling?
use Berendsen pressure bath coupling? 
Acceptable Values: on or off
Default Value: off
Description: Specifies whether or not Berendsen pressure bath coupling is active.
If set to on, then the parameters BerendsenPressureTarget, BerendsenPressureCompressibility and BerendsenPressureRelaxationTime must be set
and the parameter BerendsenPressureFreq can
optionally be set to control the behavior of this feature.
- BerendsenPressureTarget
target pressure (bar)
Acceptable Values: positive decimal
Description: Specifies target pressure for Berendsen's method.
A typical value would be 1.01325 bar, atmospheric pressure at sea level.
- BerendsenPressureCompressibility
compressibility (bar
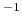 )
)
Acceptable Values: positive decimal
Description: Specifies compressibility for Berendsen's method.
A typical value would be 4.57E-5 bar
, corresponding to liquid water.
The higher the compressibility, the more volume will be adjusted for a
given pressure difference.
The compressibility and the relaxation time appear only as a ratio in the
dynamics, so a larger compressibility is equivalent to a smaller relaxation
time.
- BerendsenPressureRelaxationTime
relaxation time (fs)
Acceptable Values: positive decimal
Description: Specifies relaxation time for Berendsen's method.
If the instantaneous pressure did not fluctuate randomly during a simulation
and the compressibility estimate was exact then
the inital pressure would decay exponentially to the target pressure with
this time constant.
Having a longer relaxation time results in more averaging over pressure
measurements and hence smaller fluctuations in the cell volume.
A reasonable choice for relaxation time would be 100 fs.
The compressibility and the relaxation time appear only as a ratio in the
dynamics, so a larger compressibility is equivalent to a smaller relaxation
time.
- BerendsenPressureFreq
how often to rescale positions
Acceptable Values: positive multiple of nonbondedFrequency and fullElectFrequency
Default Value: nonbondedFrequency or fullElectFrequency if used
Description: Specifies number of timesteps between position rescalings for Berendsen's method.
Primarily to deal with multiple timestepping integrators, but also to reduce
cell volume fluctuations, cell rescalings can occur on a longer interval.
This could reasonably be between 1 and 20 timesteps, but the relaxation time
should be at least ten times larger.
NAMD provides constant pressure simulation using a modified Nosé-Hoover method in which Langevin dynamics is used to control fluctuations in the barostat.
This method should be combined with a method of temperature control, such as Langevin dynamics, in order to simulate the NPT ensemble.
The Langevin piston Nose-Hoover method in NAMD is a combination of the
Nose-Hoover constant pressure method as described in
GJ Martyna, DJ Tobias and ML Klein, "Constant pressure molecular dynamics
algorithms", J. Chem. Phys 101(5), 1994,
with piston fluctuation control implemented using Langevin dynamics as in
SE Feller, Y Zhang, RW Pastor and BR Brooks, "Constant pressure molecular
dynamics simulation: The Langevin piston method", J. Chem. Phys. 103(11),
1995.
The equations of motion are:
Here, 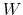 is the mass of piston,
is the mass of piston, 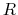 is noise on atoms, and
is noise on atoms, and 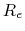 is
the noise on the piston.
is
the noise on the piston.
The user specifies the desired pressure, oscillation and decay times
of the piston, and temperature of the piston. The compressibility of
the system is not required. In addition, the user specifies the
damping coefficients and temperature of the atoms for Langevin dynamics.
The following parameters are used to define the algorithm.
- LangevinPiston
use Langevin piston pressure control?
Acceptable Values: on or off
Default Value: off
Description: Specifies whether or not Langevin piston pressure control is active.
If set to on, then the parameters LangevinPistonTarget, LangevinPistonPeriod, LangevinPistonDecay and LangevinPistonTemp must be set.
- LangevinPistonTarget
target pressure (bar)
Acceptable Values: positive decimal
Description: Specifies target pressure for Langevin piston method.
A typical value would be 1.01325 bar, atmospheric pressure at sea level.
- LangevinPistonPeriod
oscillation period (fs)
Acceptable Values: positive decimal
Description: Specifies barostat oscillation time scale for Langevin piston method.
If the instantaneous pressure did not fluctuate randomly during a simulation
and the decay time was infinite (no friction) then the cell volume would
oscillate with this angular period.
Having a longer period results in more averaging over pressure measurements
and hence slower fluctuations in the cell volume.
A reasonable choice for the piston period would be 200 fs.
- LangevinPistonDecay
damping time scale (fs)
Acceptable Values: positive decimal
Description: Specifies barostat damping time scale for Langevin piston method.
A value larger than the piston period would result in underdamped
dynamics (decaying ringing in the cell volume) while a smaller value
approaches exponential decay as in Berendsen's method above.
A smaller value also corresponds to larger random forces with increased
coupling to the Langevin temperature bath.
Typically this would be chosen equal to or smaller than the piston period,
such as 100 fs.
- LangevinPistonTemp
noise temperature (K)
Acceptable Values: positive decimal
Description: Specifies barostat noise temperature for Langevin piston method.
This should be set equal to the target temperature for the chosen method of temperature control.
- SurfaceTensionTarget
Surface tension target (dyn/cm)
Acceptable Values: decimal
Default Value: 0.0
Description: Specifies surface tension target. Must be used with
useFlexibleCell and periodic boundary conditions. The pressure
specified in LangevinPistonTarget becomes the pressure along the z
axis, and surface tension is applied in the x-y plane.
- StrainRate
initial strain rate
Acceptable Values: decimal triple (x y z)
Default Value: 0. 0. 0.
Description: Optionally specifies the initial strain rate for pressure control.
Is overridden by value read from file specified with extendedSystem.
There is typically no reason to set this parameter.
- ExcludeFromPressure
Should some atoms be excluded from pressure
rescaling?
Acceptable Values: on or off
Default Value: off
Description: Specifies whether or not to exclude some atoms from pressure rescaling. The
coordinates and velocites of such atoms are not rescaled during constant
pressure simulations, though they do contribute to the virial calculation.
May be useful for membrane protein simulation. EXPERIMENTAL.
- ExcludeFromPressureFile
File specifying excluded atoms
Acceptable Values: PDB file
Default Value: coordinates file
Description: PDB file with one column specifying which atoms to exclude from pressure
rescaling. Specify 1 for excluded and 0 for not excluded.
- ExcludeFromPressureCol
Column in PDB file for specifying
excluded atoms
Acceptable Values: O, B, X, Y, or Z
Default Value: O
Description: Specifies which column of the pdb file to check for excluded atoms.
Next: User Defined Forces
Up: Standard Minimization and Dynamics
Previous: Temperature Control and Equilibration
Contents
Index
http://www.ks.uiuc.edu/Research/namd/