

Next: Running VMD on Supercomputers
Up: VMD Tutorial
Previous: Working with Multiple Molecules
Subsections
MultiSeq (Roberts et al., BMC Bioinformatics, 7:382, 2006) is a bioinformatics analysis environment developed in the Luthey-Schulten Group at the University of Illinois in Urbana-Champaign. MultiSeq allows users to organize, display, and analyze both sequence and structure data for proteins and nucleic acids4, and has been incorporated in VMD as a plugin tool starting with VMD version 1.8.5. In this section you will learn how to compare protein structures and sequences with the VMD MultiSeq plugin. We will again use the water transporting channel protein, aquaporin, as an example.
Very often comparing structures of different proteins reveal many important information. For example, proteins with similar functions tend to be found with similar structural features. MultiSeq structure alignment is useful for this reason. We will compare the structures of four aquaporin proteins, whose coordinate files can all be found in the vmd-tutorial-files directory.
Table 8:
The four aquaporins used in this section.
| PDB code |
Description |
| 1fqy (Murata et al., Nature, 407:599, 2000) |
Human AQP1 |
| 1rc2 (Savage et al., PLoS Biology, 1:E72, 2003) |
E. coli AqpZ |
| 1lda (Tajkhorshid et al., Science, 296:525, 2002) |
E. coli Glycerol Facilitator (GlpF) |
| 1j4n (Sui et al., Nature, 414:872, 2001) |
Bovine AQP1 |
|
- 1
- Start a new VMD session. Open the Molecule File Browser window by choosing the File
 New Molecule... menu item in the VMD Main window. In the Molecule File Browser window, use the Browse... button to find and select the file 1fqy.pdb in the directory vmd-tutorial-files. Press Load to load the molecule.
New Molecule... menu item in the VMD Main window. In the Molecule File Browser window, use the Browse... button to find and select the file 1fqy.pdb in the directory vmd-tutorial-files. Press Load to load the molecule.
- 2
- Load the remaining aquaporins, 1rc2, 1lda, and 1j4n. Make sure that each PDB is loaded into a new molecule. Close the Molecule File Browser window when you finished loading all four molecules. Your VMD Main menu should look like Fig. 32 when all four aquaporins are loaded.
Figure 32:
VMD Main menu after loading the four aquaporins.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.7\textwidth]{FIGS/four_aqp}
}
\end{center}
\end{figure}](img83.gif) |
- 3
- Within the VMD main window, choose the Extensions menu and select Analysis
MultiSeq.
The Multiseq window (with window name untitled.multiseq showing on top) should now be open. You may be asked to update some databases in a pop-up window if this is
the first time you use MultiSeq. If this is the case, simply click Yes and wait for MultiSeq to finish downloading. When MultiSeq starts, your Multiseq window should look
like Fig. 33, with a list of the four aquaporin protein structures and a list of two non-protein structures. The non-protein structures are detergent molecules used in
crystallizing the aquaporin proteins, and will not be needed for structure or sequence alignment. You can tell MultiSeq to throw away molecules you are not interested in.
Figure 33:
The MultiSeq window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/multiseq}
}
\end{center}
\end{figure}](img84.gif) |
- 4
- In the Multiseq window, select the 1lda_X detergent molecule by clicking on it. This will highlight the entire row of 1lda_X. Remove it from MultiSeq by pressing the delete or Backspace key on your keyboard. Do the same to remove the 1j4n_X detergent molecule.
MultiSeq uses the program STAMP (Russell et al., Proteins: Struct., Func., Gen., 14:309, 1992) to align protein molecules. STAMP (Structural Alignment of Multiple Proteins)
is a tool for aligning protein sequences based on a three-dimensional
structure. Its algorithm minimizes the 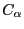 distance between aligned residues of each
molecule by applying globally optimal rigid-body rotations and translations.
Note that you can only perform alignments on molecules
that are structurally similar; if you try to align proteins that have no
common structures, STAMP will have no means of aligning them.
distance between aligned residues of each
molecule by applying globally optimal rigid-body rotations and translations.
Note that you can only perform alignments on molecules
that are structurally similar; if you try to align proteins that have no
common structures, STAMP will have no means of aligning them.
- 5
- In the Multiseq window, select Tool
Stamp Structural Alignment. This will open the Stamp Alignment Options window.
- 6
- In the Stamp Alignment Options window,
choose Align the following: All Structures and
go to the bottom of the menu and press OK.
The molecules have been aligned. You can see the alignment both in the OpenGL window and in the MultiSeq window (Fig. 34). Your alignment in OpenGL window will not immediately resemble Fig. 34. When MultiSeq completes an alignment, it creates a new representation for all the aligned protein in the NewCartoon representation with the same default coloring method and hides all other representations created previously. Let's give different colors to different aquaporins to distinguish them.
Figure 34:
The four aquaporins aligned according to their structural similarity.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/structure_aligned}
}
\end{center}
\end{figure}](img86.gif) |
- 7
- Open your Graphical Representations window, and you should see two representations for each molecule, one on top created when VMD loaded the molecule (which is now hidden), and one on the bottom created automatically by MultiSeq. Select 0:1fqy.pdb in the Selected Molecule pull-down menu on top and highlight the bottom representation by clicking on it. Change the color for this representation by selecting ColorID
1 red for Coloring Method.
- 8
- In the Graphical Representations window, select 1:1rc2.pdb in the Selected Molecule pull-down menu on top and highlight the bottom representation by clicking on it. Select ColorID
4 yellow for Coloring Method.
- 9
- In the Graphical Representations window, select 2:1lda.pdb in the Selected Molecule pull-down menu on top and highlight the bottom representation by clicking on it. Select ColorID
11 purple for Coloring Method.
- 10
- In the Graphical Representations window, select 3:1j4n.pdb in the Selected Molecule pull-down menu on top and highlight the bottom representation by clicking on it. Select ColorID
12 lime for Coloring Method. Close the Graphical Representations window.
Now your OpenGL window should look similar to Fig. 34, and you can see that the alignment was pretty good as the four aquaporin structures are very similar.
You can also get more information about the alignment in the MultiSeq window by highlighting the molecules you wish to compare.
- 11
- In the MultiSeq window, highlight 1fqy by clicking on it. To highlight another molecule without unhighlighting 1fqy, you need to Ctrl-click (or command-click on Mac) on that molecule. Highlight 1rc2 by clicking on it while holding down the Ctrl key on the keyboard (or the command key on Mac). When both 1fqy and 1rc2 are highlighted, you should see on the lower left corner in the MultiSeq window a line of text: QH:0.6442, RMSD:2.3043, Percent Identity:30.28 as shown in Fig. 35. Note, the values you obtain might be a little different depending on if your MultiSeq database is updated, but they should be close to the ones in Fig. 35.
Figure 35:
The Q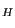 , RMSD, and Percent Identity values can be used to determine how good an alignment is between two molecules, and how
similar they are in structure and sequence.
, RMSD, and Percent Identity values can be used to determine how good an alignment is between two molecules, and how
similar they are in structure and sequence.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.4\textwidth]{FIGS/alignment_info}
}
\end{center}
\end{figure}](img87.gif) |
The Q
value is a metric for structural homology. It's an adaptation of the Q value that measures structural conservation5. Q=1 implies that structures are identical. When Q has a low score (0.1-0.3), structures are not aligned well, i.e., only a small fraction of the
atoms superimpose. Along with RMSD and Percent Identity, these numbers tell you that the 1fqy and 1rc2 structures are pretty well aligned. You can repeat the previous step to compare the alignment of other molecules. To unselect a highlighted molecule, Ctrl-click on it again (or command-click on Mac).
You can also color the molecules according to the value of Q per residue (Q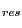 ) obtained in the alignment. Q
is the contribution from each residue to the overall Q value of aligned structures.
) obtained in the alignment. Q
is the contribution from each residue to the overall Q value of aligned structures.
- 12
- In the MultiSeq window, choose View
Coloring
Qres.
- 13
- Look at the OpenGL window to see the impact this selection has made on the coloring of the aligned molecules (Fig. 36). The blue areas indicate that the molecules are structurally conserved at those points. If there is no correspondence in structural proximities at these points, the points appear red. As you can see the
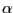 -helices that form the pore are well conserved structurally among the four aquaporins, while there are more structural difference in the less functionally relevant loops.
-helices that form the pore are well conserved structurally among the four aquaporins, while there are more structural difference in the less functionally relevant loops.
Figure 36:
Result of a structural alignment of the four aquaporins, colored by Q
.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/colored_by_qres}
}
\end{center}
\end{figure}](img88.gif) |
Besides studying structural similarities, MultiSeq also allows protein comparison based on their sequence information. Sequence alignment is often used to identify conserved residues among similar proteins, as such residues are likely functionally important.
- 1
- In the MultiSeq window, select Tools
Sequence Alignment window. In the Sequence Alignment Options window,
choose ClustalW under Alignment Program and make sure the Align All Sequences option is checked, and go to the bottom of the window and select OK. Now the four aquaporins have been aligned according to their sequence using the ClustalW tool (Thompson et al., Nucl. Acids Res., 22:4673, 1994).
- 2
- Let's color the aligned molecules by their sequence similarity. In the Multiseq window, choose View
Coloring
Sequence identity. Now each amino acid is colored according to the degree of conservation within the alignment: blue means highly conserved, whereas red means very low or no conservation. Your MultiSeq window and OpenGL window should now resemble Fig. 37.
Figure 37:
Result of a sequence alignment of the four aquaporins, colored by sequence identity.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/colored_by_seqid}
}
\end{center}
\end{figure}](img89.gif) |
You have now aligned the four aquaporins by their sequence and identified the conserved residues, which tend to locate inside the pore (Fig. 38). Since aquaporin facilitates water transport across the membrane, these conserved residues are most likely the ones that carry out this important function.
Figure 38:
Top view of the aligned aquaporins colored by sequence conservation. The conserved residues locate mostly inside the aquaporin pore.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.6\textwidth]{FIGS/seq_alignment_top}
}
\end{center}
\end{figure}](img90.gif) |
Many times the structure of a protein might not be available, but its sequence is. You can analyze a protein in MultiSeq without its structure by loading its sequence information in the FASTA file format. If you don't have the FASTA file of a protein but you have its sequence, you can create a FASTA file easily with any text editor of your choice.
- 3
- In the vmd-tutorial-files directory, find the provided FASTA sequence file spinach_aqp.fasta and open it with a text editor (Fig. 39a). A FASTA file contains a header that starts with ``
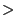 ", followed by the name of the protein. In the next line is the protein sequence in one-letter amino acid code. You can create FASTA files similarly in this format. When you create a FASTA file, remember to save it in plain text, and use .fasta as the file extension. Close the text editor when you finish examining spinach_aqp.fasta.
", followed by the name of the protein. In the next line is the protein sequence in one-letter amino acid code. You can create FASTA files similarly in this format. When you create a FASTA file, remember to save it in plain text, and use .fasta as the file extension. Close the text editor when you finish examining spinach_aqp.fasta.
Figure 39:
The content of spinach_aqp.fasta.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.9\textwidth]{FIGS/spinach_aqp}
}
\end{center}
\end{figure}](img92.gif) |
- 4
- In the MultiSeq window, select File
Import Data.... Select From File in the Import Data window, and press the top Browse button to select the file spinach_aqp.fasta. Press OK on the bottom of the Import Data window.
You have now loaded the sequence information of a spinach aquaporin into MultiSeq (Fig. 39b). You can now preform sequence alignment on the spinach aquaporin protein with other loaded aquaporin molecules. Let's try a sequence alignment between spinach and human aquaporins.
- 5
- Click on the checkbox on the left of spinach_aqp, and click on the checkbox on the left of 1fqy.pdb. Open the Sequence Alignment Options window by selecting Tools
Sequence Alignment. Choose ClustalW as the Alignment Program and under the Multiple Alignment options on the top, check Align Marked Sequences. Go to the bottom of the window and select OK.
The sequence of spinach aquaporin is now aligned with the sequence of human aquaporin, and you can check how good the alignment is by obtaining its Q
and Sequence Identity values. If you feel that the two molecules are listed too far apart in the MultiSeq window, you can move the molecules by dragging them with your mouse. Also, as you might have noticed, in MultiSeq molecules can be ``Marked'' by checking their checkboxes. They can also be ``Selected'' by highlighting them. You can align only the molecules of your choice by selecting Align Marked Sequences or Align Selected Sequences, depending if you have marked or highlighted your molecules. This option is available for both structural alignment and sequence alignment.
The structures of spinach aquaporin are actually available (Törnroth-Horsefield et al., Nature, 439:688, 2006), but now that you have learned how to import FASTA sequence data, you can compare the sequences of proteins even if their structures are not yet published.
- 6
- When you finish comparing the sequence of spinach aquaporin with other aquaporins, delete it by clicking on spinach_aqp and press delete or Backspace on your keyboard.
The Phylogenetic Tree feature in MultiSeq elucidates the structure-based and/or sequence-based relationships between different proteins. Structure-based phylogenetic trees can be constructed according to the RMSD or Q values between the
molecules after alignment; sequence-based phylogenetic trees can be constructed according to the percent identity or ClustalW values.
- 1
- Align the structures again, by going to the Multiseq
window and selecting Tools
Stamp Structural Alignment.
- 2
- In the Stamp Structural Alignment window, select All
Structures, and keep the default values for the rest of the
parameters. Press the OK button to align the structures.
- 3
- In the Multiseq program window choose Tools
Phylogenetic Tree. The Phylogenetic tree window will open.
- 4
- Select Structural tree using Q
, and
press the OK button. A phylogenetic tree based on the Q
values will be calculated and drawn as shown in
Fig. 40. Here you can see the relationship between the four
aquaporin, e.g., how the E.Coli AqpZ (1r2c) is related to human AQP1
(1fqy).
Figure 40:
a) A structure-based phylogenetic tree generated by Q
values. b) A sequence-based phylogenetic tree generated by ClustalW.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[width=0.8\textwidth]{FIGS/trees}
}
\end{center}
\end{figure}](img95.gif) |
- 5
- You can also construct the phylogenetic tree of the four aquaporins
based on their sequence information. Close the Tree Viewer window
- 6
- You need to perform the sequence alignment again for the four aquaporin proteins. In your MultiSeq window, choose Tools
Sequence Alignment, check ClustalW under Alignment Program and make sure the Align All Sequences option is checked, and press OK.
- 7
- In the Multiseq program window choose Tools
Phylogenetic Tree to open Phylogenetic tree window again.
- 8
- Select Sequence tree using ClustalW, and
press the OK button. A phylogenetic tree based on ClustalW will be calculated and drawn as shown in
Fig. 40.
- 9
- Quit VMD.
Next: Running VMD on Supercomputers
Up: VMD Tutorial
Previous: Working with Multiple Molecules
vmd@ks.uiuc.edu
![\fbox{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2.3 cm]{FIGS/tut0...
...{Inferring Phylogenies}
by Joseph Felsenstein\footnotemark .}
\end{minipage} }](img94.gif)