

Next: Ubiquitin in a Water
Up: Basics of NAMD
Previous: Solvating the Protein
Subsections
In this section, you will examine the minimization and equilibration of ubiquitin in a water sphere placed in vacuum.
- 1
- Go to your 1-2-sphere directory by typing cd ../1-2-sphere. Here, you will find a configuration file for the minimization and equilibration of ubiquitin in a water sphere.
All output files for the minimization and equilibration of your ubiquitin in a water sphere system will be placed in this directory. The configuration file is the only input file placed here, since it is particular to this simulation. The pdb, psf, and parameter files, which may be used by many other simulations, are placed in your common directory and are called by each respective configuration file.
- 2
- Open the configuration file, ubq_ws_eq.conf by typing nedit ubq_ws_eq.conf.
The configuration file may seem complex at first, but it will be examined line by line to determine its function in your simulation.
Note that when ``#" appears at the beginning of a line, the entire line is not recognized as compilable script. These lines are ``commented out", rather. In the middle of a line, ``;#" is used to comment out the remainder of the line.
- 3
- The ``Job Description" section contains only comments, and its purpose is to inform those who view the configuration file about what it is meant for. Your comment should read
| # Minimization and Equilibration of |
|
| # Ubiquitin in a Water Sphere |
|
- 4
- The ``Adjustable Parameters" section contains five commands:
- structure: calls the psf file describing the system (../common/ubq_ws.psf).
- coordinates: calls the initial coordinate data from the file listed next to it (in this case ../common/ubq_ws.pdb).
- set temperature: creates a variable called ``temperature" in which to store a value for the initial temperature of the system. If you place the text ``$temperature" in the configuration file, NAMD simply reads it as a label for the number ``310". (Creating variables is useful since you can alter the value of that variable in a single place if it is listed many times in the configuration file.)
- set outputname: creates a variable called ``outputname" in which to store a generic name for output files. If you place the text ``$outputname" in the configuration file, NAMD simply reads it as a label for ``ubq_ws_eq".
- firsttimestep: simply sets a number value for the first time step of the simulation. It is typically useful when restarting a simulation. For instance, if a previous simulation ended at time step 552, the command firsttimestep 553 would be used.
- 5
- The ``Simulation Parameters" section contains many commands, commented into different categories:
- Input
- paraTypeCharmm: indicates whether or not the parameter file is in the format used by the CHARMM force field. on indicates that it is; off indicates that it is not. (If this command is not specified, NAMD assumes the file is in X-PLOR format by default.)
- parameters: calls the force field parameters from the file listed next to it (in this case ../common/par_all27_prot_lipid.inp).
- temperature: sets the initial temperature of the system, in K, with the value listed next to it (in this case $temperature, or 310). This is done by assigning random velocities to atoms such that their average kinetic energy accurately represents the given temperature.
- Force-Field Parameters
- exclude: specifies which atomic interactions are to be excluded from consideration. See Figure 4 for general atom labels. The scaled1-4 value indicates that interactions between any such atoms 1 and 3 are neglected and interactions between atoms 1 and 4 are modified. The van der Waals interaction for ``1-4" atoms are modified using special 1-4 parameters defined in the parameter files, and electrostatic interaction is modified as shown in the next command.
Figure 4:
Number labels for atoms which are a given amount of bonds away from one another
![\begin{figure}\begin{center}
\par\par\latex{
\includegraphics[scale=0.5]{pictures/tut_unit01_bonds}
}
\end{center} \end{figure}](img29.png) |
- 1-4scaling: specifies the degree to which the electrostatic interaction between 1-4 atoms is to be taken into account. It may be a decimal between 0 and 1 (here, it is 1) and indicates how much the interaction is ``turned off" or ``on", respectively.
- cutoff: indicates the distance in Å beyond which electrostatic and van der Waals interactions are cut-off. Otherwise, those interactions are considered over the entire volume of your system. This can be computationally too costly. The definition changes when the Particle Mesh Ewald Sum is invoked. See Section 1.5.1.
- switching: indicates whether or not switching functions are used to smoothly take electrostatic and van der Waals interactions to zero at the cutoff distance. You may list ``on" or ``off" next to it as a yes/no answer.
- switchdist: indicates the distance in Å at which the functional form of electrostatic and van der Waals interactions is modified to allow their values to approach zero at the cutoff distance. A visual explanation of this is useful and may be found in Figure 5.
- pairlistdist: is designed to make computation faster. It specifies a distance in Å. NAMD will only search within this distance for atoms which may interact by electrostatic or van der Waals interactions. This way, NAMD does not have to search the entire system. The distance must be greater than the cutoff distance, and the list must be updated during the simulation. See Figure 5.
Figure 5:
Cutoff and switching distances indicated on the left. Pair list distance indicated on the right.
![\begin{figure}\begin{center}
\par\par\latex{
\includegraphics[scale=0.5]{pictures/tut_unit01_cutoff}
}
\end{center} \end{figure}](img30.png) |
- Integrator Parameters
- timestep: indicates the value of the time step size used in the simulation. MD simulations solve Newton's laws to determine the trajectories of atoms. The time step tells NAMD how to discretize the particle dynamics. It is specified in femtoseconds (here, 2 fs).
- rigidBonds: specifies which bonds involving hydrogen are considered to be rigid (non-vibrating). The value all specifies all linear bonds involving hydrogen and any other atoms.
- nonbondedFreq: specifies in number of time steps how often nonbonded interactions should be calculated. It is useful for saving computational time.
- fullElectFrequency: specifies in number of time steps how often full electrostatic interactions should be calculated.
- stepspercycle: Atoms are reassigned pair list identities (as explained above) once every cycle. This command specifies how long one cycle lasts, i.e. the number of time steps in one cycle.
- Constant Temperature Control
- langevin: indicates whether or not the simulation uses Langevin dynamics; uses values on and off. See the science box below for more on Langevin dynamics.
- langevinDamping: sets the value of the Langevin coupling coefficient, which quantifies the friction applied to the system, removing energy from the system, slowing atoms down, etc. It is specified in ps
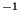 .
.
- langevinTemp: Langevin dynamics may be applied to all atoms or only non-hydrogen atoms in the system. This command specifies the temperature at which to keep those atoms, even though friction and random forces will be acting on them. (Remember $temperature is a variable for the value 310.)
- langevinHydrogen: indicates whether or not Langevin dynamics will be applied to hydrogen atoms in the simulation; uses values on and off.
- Output

![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...t acceptable, but should be avoided for accurate simulations.}
\end{minipage} }](img33.png)
- 6
- The ``Extra Parameters" section contains commands which are applicable to more specific simulations. Here have been placed commands which characterize the spherical boundary conditions on the water sphere. These conditions prevent the sphere from undergoing evaporation or diffusion.
- 7
- The ``Execution Script" section contains three commands, the first two of which apply to minimization and the last one of which applies to equilibration.
- Minimization
- minimize: sets the number of iterations over which to vary atom positions to search for a minimum in the potential (in this case 100).
- reinitvels: Minimization is performed on the system after all atomic velocities have been set to zero. This command resets atomic velocities such that the system starts at the temperature specified (in this case, $temperature, or 310K).
- run: sets the number of time steps over which to run the MD equilibration (in this case 5000, which corresponds to 10,000 fs or 10 ps, since a 2 fs time step has been used).
- 8
- Now, close the configuration file by clicking File
 Exit.
Exit.
Run your Simulation
Your simulation should take about 20 minutes to complete, and NAMD will produce the output files described in Section 1.6.
Next: Ubiquitin in a Water
Up: Basics of NAMD
Previous: Solvating the Protein
namd@ks.uiuc.edu

![% latex2html id marker 3324
\framebox[\textwidth]{
\begin{minipage}{.2\textwidt...
...rrent/ug/}{http://www.ks.uiuc.edu/Research/namd/current/ug/}.}
\end{minipage} }](img36.png)
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...owing: {\tt temperature, velocities,} or {\tt binvelocities}.}
\end{minipage} }](img37.png)
