From: Ishaan Roy (royi_at_uchicago.edu)
Date: Thu Aug 26 2021 - 02:28:47 CDT
Hi Peter,
Nothing stands out as weird about the runs, visually or otherwise. The sampling distribution is, I believe, as one would expect, generally increasing for higher values of the coordinate and the trajectory follows a square-root diffusion trend. There’s no sign of the protein getting stuck anywhere.
Thanks,
Ishaan
From: Peter Freddolino<mailto:petefred_at_umich.edu>
Sent: Wednesday, August 25, 2021 10:34 PM
To: <namd-l_at_ks.uiuc.edu><mailto:namd-l_at_ks.uiuc.edu>; Ishaan Roy<mailto:royi_at_uchicago.edu>
Subject: Re: namd-l: Unrealistically High Keq in eABF Seperation
Dear Ishaan,
If your simulations haven't yet converged, there is really no sense in evaluating the energies completely yet. With that said, how is the sampling looking across the different values of your reaction coordinate? Is the reaction coordinate stuck somewhere or is the system diffusing freely along it?
Have you visually inspected your system to make sure nothing weird is happening?
Best,
Peter
On Tue, Aug 24, 2021 at 7:08 PM Ishaan Roy <royi_at_uchicago.edu<mailto:royi_at_uchicago.edu>> wrote:
Hello,
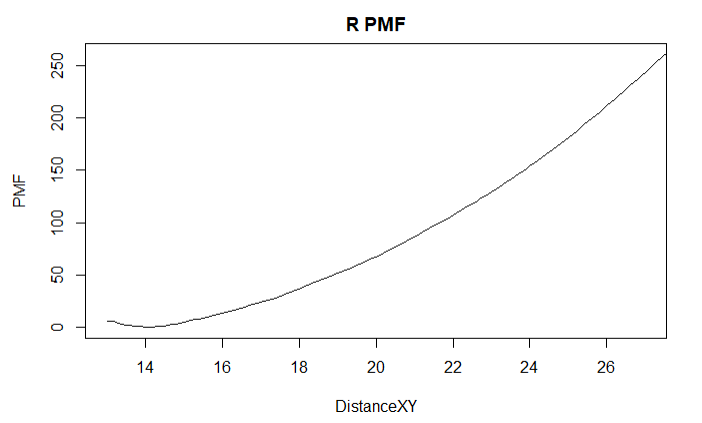
I am using eABF to separate two membrane proteins to find the free binding energy. The PMF values produced in my runs seem to be incredibly high compared to consistently reaching the 100s order of magnitude. We haven’t completed a run that plateaus in free energy yet, but this is an example showing what the PMF looks like.
[cid:17b807eaccd56cdf7d61]
A maximum PMF this high already produces a value for the binding constant that is on an order beyond 10^100. Besides being high, it might be worth mentioning that the nonbonded energies are very close to 0 near the end of this run, but the PMF gradient doesn’t level off at all. Any ideas on what might be causing this? Or is such a value plausible? This only seems to be a problem with the separation runs; my other colvars produce PMF plots that are closer to what I’ve seen.
Thanks,
Ishaan
This archive was generated by hypermail 2.1.6 : Fri Dec 31 2021 - 23:17:11 CST