From: Rabeta Yeasmin (rabetayeasmin_at_gmail.com)
Date: Tue Jul 10 2018 - 09:46:07 CDT
Hi Freddolino,
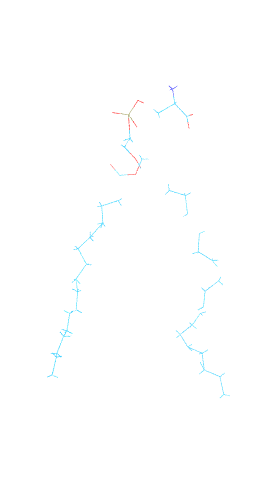
Actually the atoms were in the approximately right places, but they are not
bonded in some place. If you take a look at the attached lipid figure, you
will see that the tails and head of the lipid are not connected in some
places. But after running the reveterted for a while, the problem looks
like solved.
Rabeta Yeasmin
On Tue, Jul 10, 2018 at 9:07 AM, Peter Freddolino <petefred_at_umich.edu>
wrote:
> Can you please elaborate on what you mean by "broken"? The way that namd
> does reverse CG should give structures that have atoms in roughly the right
> places, but the orientations will not be perfect, and some relaxation will
> be required. Quoting the cgtools documentation:
> "A simulated annealing run from NAMD will usually need to be run after
> reconstruction of the all-atom model. The annealing run needs to be run in
> a specific way, so the CG builder tool can create the proper NAMD
> configuration files to use. By default the CHARMM parameter file (used by
> several other VMD plugins) will be used for the config file, but you can
> alter this as desired. In addition, the PSF filename will be needed for the
> NAMD simulation."
>
> The config file generated by cgtools should have appropriate constraints
> to run the needed refinement.
> Best,
> Peter
>
> On Mon, Jul 9, 2018 at 1:38 PM, Rabeta Yeasmin <rabetayeasmin_at_gmail.com>
> wrote:
>
>> Hi,
>>
>> I have a protein-lipid system that I converted to a coarse-grained system
>> using residue-based coarse-graining method and run Steered MD and
>> umbrella sampling. After running around 350ns, I have reverted the last
>> structure of all windows to the all-atom system using the
>> reverse-residue-based coarse-gaining method. But I found the protein and
>> lipid structure are broken in the all-atom system. Is that am a usual
>> thing?
>> Thanks.
>>
>> Rabeta Yeasmin
>>
>
>
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2018 - 23:21:16 CST