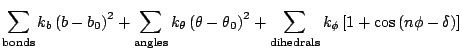
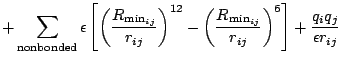
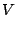

From: Yaxin An (yxan2016_at_gmail.com)
Date: Fri Jun 09 2017 - 10:38:33 CDT
Thanks, Peter. It's my first time using the namd mailist.
The model is not published. I am an MD beginner. I will give more
information about the model later. Thanks for your patience.
Bests;
Yaxin
On Fri, Jun 9, 2017 at 11:27 AM, Peter Freddolino <petefred_at_umich.edu>
wrote:
> Hi Yaxin,
> Please keep your replies on list (keep copying namd-l) so that others can
> follow the conversation. And this is not very informative, because you're
> giving a potential form used by every MD simulation ever. Is this a
> published CG model or something in house? What is the level of coarse
> graining? How stiff are the potentials? That will help to determine what
> the fastest degrees of freedom in the system are.
> Best,
> Peter
>
>
>
> On 06/09/2017 11:23 AM, Yaxin An wrote:
>
> Hi Peter
>
> Thanks for the kind reply.
>
> The potentials I am using is following
>
>
> [image: Inline image 5]
> [image: Inline image 3]
> [image: Inline image 2]
> [image: Inline image 1]
>
>
> Bests
>
> Yaxin
>
> On Fri, Jun 9, 2017 at 11:10 AM, Peter Freddolino <petefred_at_umich.edu>
> wrote:
>
>> Following up on that, it would be helpful to know what type of CG model
>> you're using and what your other parameters look like...
>>
>>
>>
>> On 06/09/2017 10:45 AM, Richard Overstreet wrote:
>>
>>> See here:
>>>
>>> http://www.ks.uiuc.edu/Research/namd/mailing_list/namd-l.201
>>> 5-2016/2439.html
>>>
>>> and
>>>
>>> http://www.ks.uiuc.edu/Research/namd/mailing_list/namd-l.201
>>> 5-2016/2431.html
>>>
>>> In brief the time step will affect the accuracy of your integrator and
>>> you are limited in the choice by the fastest degree of freedom in your
>>> simulation.
>>>
>>> On 06/09/2017 09:39 AM, Yaxin An wrote:
>>>
>>>> Hi
>>>>
>>>> I am running coarse-grained simulations with a time step of 5fs. When
>>>> the system sizes increased from 100 molecules to 500 or 1000 molecules, the
>>>> simulations crushed after running for hundreds of steps. The errors are
>>>> "atoms moving too fast" or "low global exclusion". When I used a timestep
>>>> of 10fs, these errors emerged after running for hundreds or about one
>>>> thousand steps.
>>>>
>>>> I have to restart the simulations, but after some time they crushed
>>>> again.
>>>>
>>>> Is there someone who knows how to fix this problem?
>>>>
>>>> Thanks
>>>>
>>>> Bests;
>>>> Yaxin
>>>>
>>>
>>>
>>
>
>
This archive was generated by hypermail 2.1.6 : Sun Dec 31 2017 - 23:21:21 CST