From: Francesco Pietra (chiendarret_at_gmail.com)
Date: Fri Mar 11 2011 - 11:46:30 CST
Hi Jim:
Sorry, I forgot to change to jpg format, which is now attached
francesco
On Fri, Mar 11, 2011 at 5:40 PM, Jim Phillips <jim_at_ks.uiuc.edu> wrote:
> Hi,
>
> I'm sorry, this message is too large to distribute on NAMD-L. Please do not
> attach large files to your messages. Instead, include only the relevant
> parts of the files and provide a URL (pointing to your own web server) where
> the complete files are available.
>
> Thanks!
>
> -Jim
>
>
> Date: Fri, 11 Mar 2011 16:07:20 +0100
> Message-ID: <AANLkTimK88diJ02J7MYvn3xXrWw8kL3XZk9mEN_DRxSj_at_mail.gmail.com>
> Subject: Re: Fwd: vmd-l: Re: namd-l: hBond colvars and patching
> From: Francesco Pietra <chiendarret_at_gmail.com>
> To: Peter Freddolino <pfreddol_at_princeton.edu>
> Cc: NAMD <namd-l_at_ks.uiuc.edu>, vmd-l_at_ks.uiuc.edu
> Content-Type: multipart/mixed; boundary=90e6ba4fbfe095f97c049e36514a
>
> --90e6ba4fbfe095f97c049e36514a
> Content-Type: text/plain; charset=UTF-8
> Content-Transfer-Encoding: quoted-printable
>
> Hello Peter:
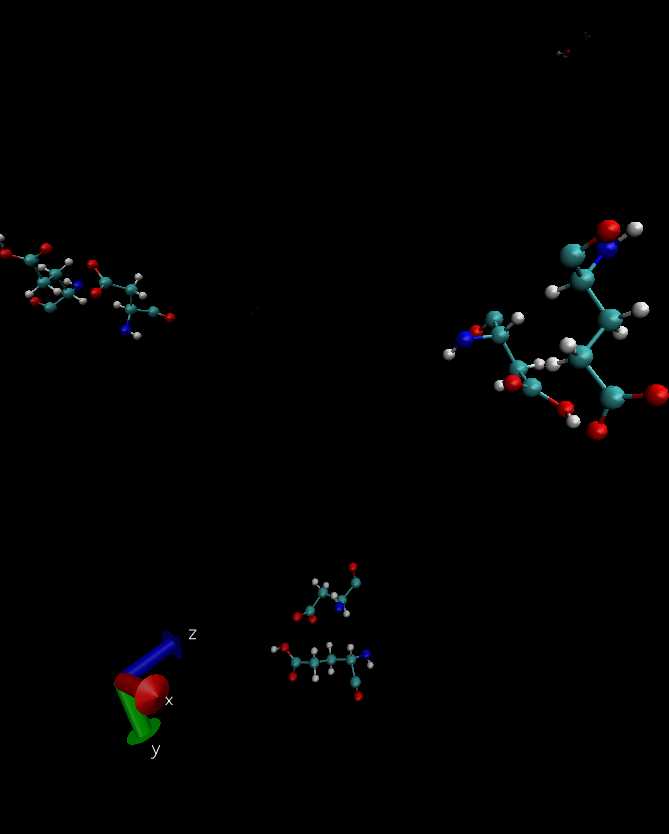
> Done as you suggested. Now the stereochemistry in the couples
> ASP-patchedGLU, GLU-patchedASP, and CLA-patchedGLU is substantially
> the same before and after unrestricted minimization in a POPC membrane
> (0 K, ts=3D1, "rigidBonds water", gradient 5.0) and seem palatable. See
> attached examples. I hope the house is in order now.
>
> I found no suggestion from the archives to decide which is best
> appropriate, "distance" or "hBond" colvars, to restrict distances from
> CLA to its ligand atoms (from patchedGLU, LYS, and ARG). Could that be
> suggested? In fact, if the are problems about that, they will only
> emerge from long simulations. My aim is to prevent CLA escaping, while
> allowing formation of the H-bond during equilibration. To this
> concern, it seems to me better not to restrain the GLU-ASP distance or
> hBond, and see what the ff feels. If it is suitable to the system, it
> is a way to see if the H-bond distances seen in the crystal hold for
> the solution state as well in this forced "constant low pH"
> simulation.
>
> Thousand thanks
> francesco
>
>
> On Fri, Mar 11, 2011 at 5:22 AM, Peter Freddolino
> <pfreddol_at_princeton.edu> wrote:
>>
>> Hi Francesco,
>> Could you try building your structure with "Regenerate angles/dihedrals"
>> (under the autopsf Options menu) selected? That should get rid of the
>> odd location of those hydrogens; in some cases psfgen fails to properly
>> add all angle terms that should be present during patching, which would
>> lead to what you saw. Also, the TIP3 topology listed below appears
>> incorrect, as it gets rid of *all* of the bonds. You only need to get
>> rid of the H1-H2 bond, which means deleting the last two words of the
>> BOND line (H1 H2) but uncommenting it.
>
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:19:55 CST