From: yun luo (luoyun724_at_gmail.com)
Date: Tue May 05 2009 - 12:19:05 CDT
Hi,
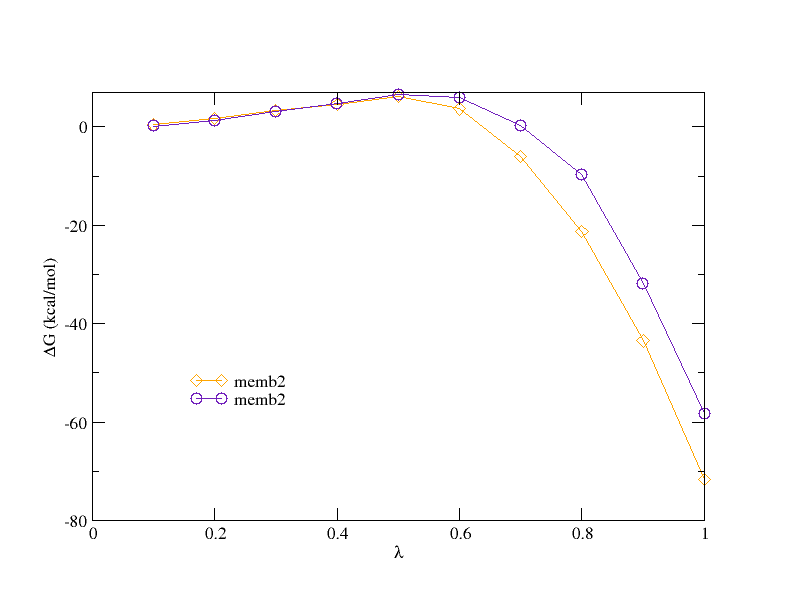
In case of disappearing a sodium ion in membrane, the VdW interaction is
much smaller than Electrostatic interaction. How to set the better values
of fepVdWLambdaEND and fepEleLambdaStart ? I use the default value 0.5 for
both parameters, the convergence of two 200ps runs seems not very well
during Elec part Lambda 0.5 to 1 (see attached figure).
On Tue, May 5, 2009 at 11:49 AM, yun luo <luoyun724_at_gmail.com> wrote:
> However, in the NAMD2.7 UserGuide, both fepVdWLambdaEND and
> fepEleLambdaStart have default value 0.5. So I guess if we don't specify
> those two value, the decoupling or coupling of VdW interaction will only be
> carried out from lambda 0 to 0.5, and decoupling or coupling of
> Electrostatic interaction will only be carried out during lambda 0.5 to 1.
> Is that right?
>
>
> On Tue, May 5, 2009 at 10:43 AM, Jerome Henin <jhenin_at_cmm.chem.upenn.edu>wrote:
>
>> Hi all,
>>
>> Since this issue has been confusing to many people, here is a figure
>> illustrating two common types of calculations, and what happens to the
>> various coupling/decoupling parameters.
>>
>> Cheers,
>> Jerome
>>
>>
>> On Tue, May 5, 2009 at 11:18 AM, daniel aguayo <bioquimico_at_gmail.com>
>> wrote:
>> > Tks Chris for your answer. I now understand the meaning of it.
>> >
>> > Best
>> >
>> > Daniel Aguayo V.
>> >
>> > On Mon, May 4, 2009 at 11:24 PM, Chris Harrison <char_at_ks.uiuc.edu>
>> wrote:
>> >>
>> >> Daniel,
>> >>
>> >> fepVdwLambdaEnd is the lambda value at which the coupling/decoupling of
>> >> vdW interactions is desired to be completed. The default is 1.0, so
>> the vdW
>> >> interactions of annihilated atoms are gradually decoupled from a system
>> from
>> >> lambda=0 to lambda=1. In complimentary fashion, the vdW interactions
>> of
>> >> appearing atoms are gradually coupled to a system from lambda=0 to
>> >> lambda=1.
>> >>
>> >> fepElecLambdaStart in a similar fashion controls the electrostatic
>> >> coupling/decoupling. The default is 0.5, which results in the
>> electrostatic
>> >> interactions of annihilated atoms being gradually attenuated from
>> lambda=0
>> >> to lambda=0.5 as they are decoupled from the system, while
>> electrostatic
>> >> interactions involving appearing atoms are gradually coupled to the
>> system
>> >> from lambda 0.5 to 1.0.
>> >>
>> >> A value of 0.5 is perhaps not the best example to use, but does this
>> >> answer your question?
>> >>
>> >>
>> >> C.
>> >>
>> >>
>> >> --
>> >> Chris Harrison, Ph.D.
>> >> Theoretical and Computational Biophysics Group
>> >> NIH Resource for Macromolecular Modeling and Bioinformatics
>> >> Beckman Institute for Advanced Science and Technology
>> >> University of Illinois, 405 N. Mathews Ave., Urbana, IL 61801
>> >>
>> >> char_at_ks.uiuc.edu Voice: 217-244-1733
>> >> http://www.ks.uiuc.edu/~char <http://www.ks.uiuc.edu/%7Echar>
>> Fax: 217-244-6078
>> >>
>> >>
>> >>
>> >> On Mon, May 4, 2009 at 7:53 PM, daniel aguayo <bioquimico_at_gmail.com>
>> >> wrote:
>> >>>
>> >>> Hi Jerome, can you explain more on the use of this new parameters
>> >>> fepElecLambdaStart and fepVdwLambdaEnd
>> >>>
>> >>> Tks
>> >>>
>> >>> Daniel Aguayo V.
>> >>> CBSM UTAL
>> >>> Chile
>> >>>
>> >>>
>> >>> On Mon, May 4, 2009 at 6:39 PM, Jerome Henin <
>> jhenin_at_cmm.chem.upenn.edu>
>> >>> wrote:
>> >>>>
>> >>>> Hi,
>> >>>> As you said, the only reason why the dummy atom is needed is because
>> >>>> NAMD 2.6 does not have soft-core potentials. The purpose of the
>> >>>> tutorial is somewhat academic, in that the "charging free energy" is
>> >>>> only meaningful within some theories of ion solvation, and is not an
>> >>>> experimental observable.
>> >>>> If you want a complete solvation free energy, then it is not
>> necessary
>> >>>> to follow the tutorial's "pseudo-single topology" approach.
>> >>>>
>> >>>> Note that if for some reason, you do want to use a dummy atom, its
>> >>>> mass will not affect the thermodynamics of the system. It should
>> >>>> typically not be less than 1.0, otherwise you may need to use smaller
>> >>>> timesteps to preserve the stability of the simulation.
>> >>>>
>> >>>> One more remark: even the charging free energy can now be computed
>> >>>> without the help of a dummy atom, since NAMD 2.7b1 allows for the
>> >>>> separate decoupling of electrostatic and L-J interactions, through
>> the
>> >>>> fepElecLambdaStart and fepVdwLambdaEnd parameters.
>> >>>>
>> >>>> Best,
>> >>>> Jerome
>> >>>>
>> >>>> On Mon, May 4, 2009 at 5:24 PM, yun luo <luoyun724_at_gmail.com> wrote:
>> >>>> > Hi Chris,
>> >>>> >
>> >>>> > Thank you for your reply.
>> >>>> > Actually, I did follow the FEP tutorial part 2. Charging a
>> spherical
>> >>>> > ion
>> >>>> > using dual-topology paradigm. That's why I want add a dummy atom.
>> In
>> >>>> > the
>> >>>> > tutorial one adds a dummy atom with 0 charge but the same radius as
>> >>>> > sodium
>> >>>> > because there is no soft-core contribution in NAMD2.6. Since
>> NAMD2.7
>> >>>> > has
>> >>>> > soft-core part, I think I need using a dummy atom with 0 charge 0
>> >>>> > radius to
>> >>>> > get both elec and vdW energy. But do you think the a dummy atom
>> with
>> >>>> > nonzero
>> >>>> > mass will cause problem?
>> >>>> >
>> >>>> > Many thanks!
>> >>>> >
>> >>>> > On Mon, May 4, 2009 at 3:33 PM, Chris Harrison <char_at_ks.uiuc.edu>
>> >>>> > wrote:
>> >>>> >>
>> >>>> >> Ly,
>> >>>> >>
>> >>>> >> With 0 charge, 0 mass, and 0 radius the dummy particle is
>> effectively
>> >>>> >> the
>> >>>> >> same as no atom .... which is of course the desired effect
>> usually.
>> >>>> >> Assuming you're not doing something unusual, then you don't need
>> the
>> >>>> >> dummy
>> >>>> >> particle. You should be able to just make the Na disappear.
>> >>>> >>
>> >>>> >> The FEP tutorial may be of help in setting up calculations. This
>> >>>> >> tutorial
>> >>>> >> is for NAMD 2.6 but the system setup process should be the same as
>> >>>> >> NAMD2.7b1.
>> >>>> >>
>> >>>> >> NAMD2.6 FEP tutorial:
>> >>>> >>
>> >>>> >>
>> http://www.ks.uiuc.edu/Research/namd/tutorial/fep/AlchemicalFEP-Mar2008.pdf
>> >>>> >>
>> >>>> >> Required files for tutorial:
>> >>>> >>
>> >>>> >>
>> >>>> >>
>> http://www.ks.uiuc.edu/Research/namd/tutorial/fep/AlchemicalFEP-Mar2008.zip
>> >>>> >>
>> >>>> >>
>> >>>> >> C.
>> >>>> >>
>> >>>> >>
>> >>>> >> --
>> >>>> >> Chris Harrison, Ph.D.
>> >>>> >> Theoretical and Computational Biophysics Group
>> >>>> >> NIH Resource for Macromolecular Modeling and Bioinformatics
>> >>>> >> Beckman Institute for Advanced Science and Technology
>> >>>> >> University of Illinois, 405 N. Mathews Ave., Urbana, IL 61801
>> >>>> >>
>> >>>> >> char_at_ks.uiuc.edu Voice: 217-244-1733
>> >>>> >> http://www.ks.uiuc.edu/~char <http://www.ks.uiuc.edu/%7Echar>
>> Fax: 217-244-6078
>> >>>> >>
>> >>>> >>
>> >>>> >>
>> >>>> >> On Mon, May 4, 2009 at 2:54 PM, yun luo <luoyun724_at_gmail.com>
>> wrote:
>> >>>> >>>
>> >>>> >>> Hi,
>> >>>> >>>
>> >>>> >>> I'm using NAMD2.7 for running FEP. I need to disappear a sodium
>> ion
>> >>>> >>> in my
>> >>>> >>> membrane. So I overlay a dummy atom with 0 mass 0 charge 0 radius
>> on
>> >>>> >>> a
>> >>>> >>> sodium ion. But I got a warning below:
>> >>>> >>>
>> >>>> >>> Warning: FOUND 1 ATOMS WITH ZERO OR NEGATIVE MASSES! CHANGED TO
>> >>>> >>> 0.001
>> >>>> >>>
>> >>>> >>> I'm wondering if the nonzero mass will effect the energy? If yes,
>> >>>> >>> how to
>> >>>> >>> stop this automatic changing?
>> >>>> >>>
>> >>>> >>> Thanks!
>> >>>> >>>
>> >>>> >>> Ly
>> >>>> >>
>> >>>> >
>> >>>> >
>> >>>>
>> >>>
>> >>>
>> >>>
>> >>> --
>> >>> saludos desde el fin del mundo
>> >>
>> >
>> >
>> >
>> > --
>> > saludos desde el fin del mundo
>> >
>>
>
>
This archive was generated by hypermail 2.1.6 : Wed Feb 29 2012 - 15:52:44 CST