




Next: Minimal Examples
Up: Constant-pH Simulations 1
Previous: Implementation Details
Contents
Index
Subsections
The constant-pH implementation is largely implemented in Tcl and can be found
in /lib/namdcph/namdcph.tcl, where the base directory is the NAMD
source home directory.
When that file has been loaded with a suitable source command, the
following commands and keywords are available and appear to the user in a way
similar to NAMD syntax.
The most significant change from normal NAMD usage is that there is generally
no need to use the run command.
One should instead use the new cphRun command;
this can only be used once per script for now.
NB, all commands and keywords are currently case sensitive!
cphRun  Run constant-pH MD
Run constant-pH MD 
Arguments:
numsteps
 numcycles
numcycles![$ ]$](img23.png)
Defaults: numcycles = 1
Description:
Execute numcycles cycles of constant-pH MD with the current settings.
Each cycle consists of 1) a neMD/MC move in both configuration and protonation
space and 2) MD based sampling in configuration space.
By default, configuration space sampling simply consists of numsteps
dynamics, as in conventional MD.
The nature of the neMD/MC moves, however, is more elaborate and controlled by
other keywords, many of which are required (see below).
- cphSetResidueState
Set the initial state of one or more titratable residues.
Acceptable Values:
segid
:
resid
:
resname
state
...
Description:
Initial residue states can be assigned in three ways (in descending order of
precedence): 1) via this command, 2) from a cphRestartFile, and 3)
randomly from the assigned pH and the current inherent pKa of each
residue.
- cphSetResiduepKai
Set the inherent pKa of one or more titratable residues.
Acceptable Values:
segid
:
resid
:
resname
pKai
...
Description:
The two step inherent pKa algorithm implemented here permits on-the-fly update
of an estimate for the pKa(s) of each residue.
These can either be guessed at the outset (the default is to use the reference
pKa) or updated as the simulation progresses.
A more accurate estimate of the inherent pKa increases the statistical
efficiency of the method, but the long time result is formally unbiased
regardless of the value.
If an extremely large or extremely small value is assigned, then the residue
will be assigned the most probable protonation state at the given pH and
likely remain fixed in that state.
- cphExcludeResidue
Exclude one or more residues from being titratable
Acceptable Values:
segid
:
resid
:
resname
...
Description:
By default, any residue that matches a titratable residue type will be allowed
to change protonation state.
This command permits specific residues to be excluded from consideration in a
manner that is similar to assigning an extreme inherent pKa (see
cphSetResiduepKai).
The main differences are that 1) the protonation state will not be modified and
remain as it is in the original PSF and 2) the protons in the residue will
not be tracked in the cphlog file.
This command is not always recommended, but is currently necessary for handling
disulfide linkages.
- cphRestartFile
Restart file for constant-pH
Acceptable Values: filename
Description:
Constant pH requires additional checkpoint information regarding the state of
the titratable residues and the nature of the neMD/MC moves.
This (optional) information is read from the file specified here.
After/during a simulation, this information is written to
[outputname].cphrst.
- cphRestartFreq
Frequency at which constant-pH checkpoint files are written
Acceptable Values: Non-negative integer
Default Value: 0
Description:
Checkpoint information is written to [outputname].cphrst every
cphRestartFreq cycles (not MD steps).
A checkpoint file is always written at the end of the last cycle.
- cphOutFile
Log file for constant-pH
Acceptable Values: filename
Default Value: [outputname].cphlog
Description: Titratable residue state information is logged here after every cycle.
- cphProposalWeight
MC move label and weight specifications
Acceptable Values:
move label
weight
move label
weight
...
Description:
During each cycle, MC moves are selected from the move set and then
accepted/rejected according to a Metropolis criterion based on the combined
inherent pKa information and pH.
The move weight affects the probability that such a move is selected.
Note that this does not affect the probability that any given proposal
is accepted, it merely increases the number of attempts at the given
proposal.
This may be useful in a system where one desires specific attention on a given
process, such as proton transfer or the exchange of a given residue, but one
does not want to assume that all other residue protonation states are
nominally fixed.
By default all moves are assigned equal weights of 1.0.
During the simulation these are automatically normalized to a discrete
probability mass function.
- cphMaxProposalAttempts
Maximum number of switch proposal attempts per cycle
Acceptable Values: integer
Default Value: 0
Description:
During each cycle, MC moves are selected from the move set and then
accepted/rejected according to a Metropolis criterion based on the combined
inherent pKa information and pH.
This process stops when either a switch move is accepted or a maximum limit is
reached.
Any value less than one defaults to the number of titratable residues in the
system.
- cphNumMinSteps
Number of steps of minimization before dynamics
Acceptable Values: integer
Default Value: 0
Description:
This is a replacement for the normal minimize command, which is not compatible
with constant-pH due to PSF modifications during initialization.
Setting this option to a modest number (100-200, say) might be necessary when
randomizing protonation states based on pH, since in that case it cannot be
assumed that the starting structure is representative of the initial
protonation state.
- cphForceConstant
force constant for alchemical switches (in kcal/mol-Å
 )
)
Acceptable Values: Non-negative decimal
Default Value: 100.0
Description:
During ``dual-topology'' alchemical switches, a harmonic bond is formed between
analogous atoms in each alchemical region.
This rigorously leaves all static thermodynamic quantities intact and is
generally expected to improve the stability of dynamic quantities.
- cphMDBasename
basename of intermediate files for equilibrium MD
Acceptable Values: string
Default Value: namdcph.md
Description:
PSF/coordinate modifications are currently done via the file system and utilize
intermediate files.
It may be advantageous to direct this I/O to a fast temporary directory.
- cphSWBasename
basename of intermediate files for nonequilibrium (switch) MD
Acceptable Values: string
Default Value: namdcph.sw
Description:
PSF/coordinate modifications are currently done via the file system and utilize
intermediate files.
It may be advantageous to direct this I/O to a fast temporary directory.
- cphUseGPU
use GPU-resident mode for dynamics calculations?
Acceptable Values: on or off
Default Value: off
Description:
Specifies whether or not to use GPU-resident mode for executing
the nonequilibrium switch and the equilibrium MD.
If on, these tasks are executed using GPU-resident dynamics,
which improves performance significantly.
If off (default), all calculations are performed on the CPU.
When cphUseGPU is on, the constant-pH scripts make use of
the following environment variables as shown below:
- export CONFIG_FILE start.namd
- export CPU_PATH=/path/to/cpu-only-build/namd3
- export GPU_PATH=/path/to/gpu-resident-build/namd3
- export CATDCD_PATH=/path/to/catdcd
- export NUM_THREADS=$SLURM_CPUS_PER_TASK
where start.namd refers to the main configuration file and
NUM_THREADS is to be the +p argument for launching NAMD.
The catdcd program comes with VMD.
The alternative to defining these environment variables is to
hardcode path names into the namdcph.core.tcl file.
Two different builds of NAMD are needed for GPU-accelerated constant-pH
to work, a CPU-only build and a GPU-resident build.
The workflow starts the calculation using the CPU-only build, e.g.,
- $CPU_PATH +p$NUM_THREADS +setcpuaffinity $CONFIG_FILE
With cphUseGPU on,
the scripts launch GPU-resident NAMD to run as a subprocess.
To maintain output compatibility with the original constant-pH
implementation, catdcd is used to glue DCD frames generated
by the NAMD subprocess. Performance can be very fast, but is
sensitive to the number of cores used for launching NAMD and
degrades quickly when attempting to schedule with too many cores.
The recommendation is to benchmark starting with a single core
(NUM_THREADS=1)
and to compare equilibrium MD performance against a similarly sized
system to determine an optimal core count.
Presently, this implementation supports only single-GPU runs.
Interested users are encouraged to look at the example
in directory lib/namdcph/examples/bbl/.
- cphNumEquilMDSteps
number of steps of low-temperature equilibration dynamics
Acceptable Values: integer
Default Value: 0
Description:
When cphUseGPU is on, the cphNumEquilMDSteps parameter may be used
instead of the minimize command (cphNumMinSteps).
Specifying cphNumEquilMDSteps
will implement low-temperature equilibration with a 0.1fs timestep.
Although GPU-resident dynamics is very fast, generally more steps
are required than when using the minimization protocol.
- GalvaniOffset
offset potential (in mV)
Acceptable Values: integer
Default Value: 0
Description:
For complex systems such as membrane proteins,
where the content of the simulation box
includes large nonaqueous regions,
the magnitude of the Galvani potential of the bulk water phase
can depart considerably from 0 mV.
By introducing an offset potential correction (
 ),
the Galvani potential of the bulk phase may be restored to 0 mV.
With the correction, the work calculated from a nonequilibrium switch becomes
),
the Galvani potential of the bulk phase may be restored to 0 mV.
With the correction, the work calculated from a nonequilibrium switch becomes
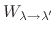

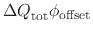 ,
where
,
where
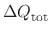 is the total charge increment resulting
from the change in protonation state.
An appropriate value of this offset parameter must be carefully determined
that is specific to the system of interest.
The user is referred to Bignucolo, et al., for
further details [8].
is the total charge increment resulting
from the change in protonation state.
An appropriate value of this offset parameter must be carefully determined
that is specific to the system of interest.
The user is referred to Bignucolo, et al., for
further details [8].
![\fbox{
\begin{minipage}[ht!]{15.3cm}
\addtolength{\baselineskip}{0.225\baselin...
...gorous of testing and may be prone
to unintended consequences.
\end{minipage}}](img422.png)
Next: Minimal Examples
Up: Constant-pH Simulations 1
Previous: Implementation Details
Contents
Index
http://www.ks.uiuc.edu/Research/namd/