Even though molecular mechanics (MM) force-fields are based on quantum mechanical calculations and experimental observations, only quantum mechanics (QM) can give a complete and accurate understanding of many biochemical processes, particularly those involving chemical reactions or charge redistribution. Nevertheless, even with the advanced hardware technology available today, the computational cost of studying nanosecond-long dynamics of entire systems relying solely on QM methodologies is usually prohibitive. A common route to circumvent this cost barrier is to confine the QM formalism to a sub-region of a system and to include the effects of the surrounding system through MM simulations, leading to hybrid QM/MM simulations [97].
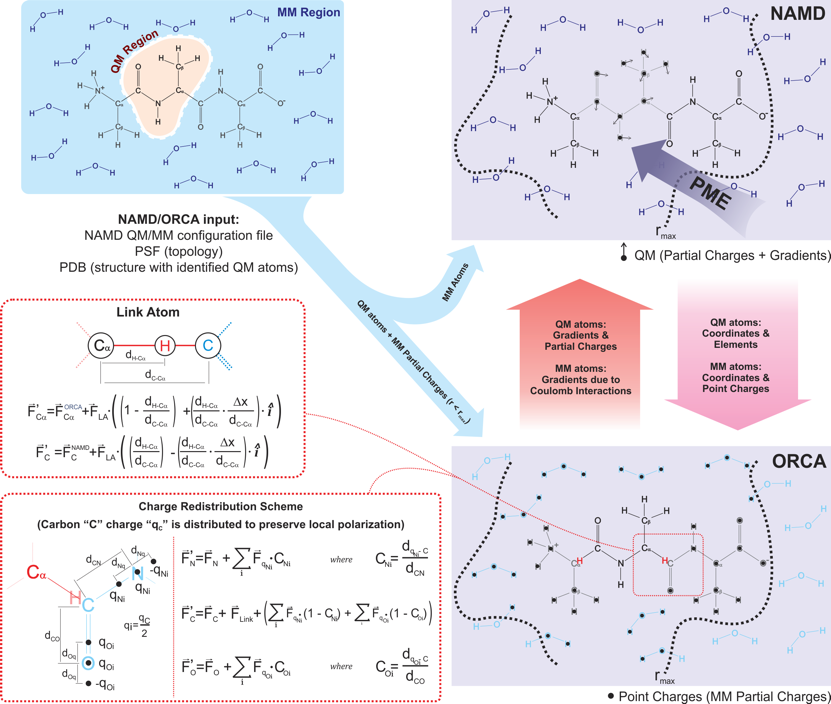
NAMD's comprehensive QM/MM suite [75] was developed to provide easy setup, visualization and analysis of QM/MM simulations through the graphical user interface VMD/QwikMD [91], and a broad range of QM methods through NAMD's new ``QMForces" module. The QM/MM interface in NAMD supports the simulation of many independent QM regions, and smooth integration with a vast collection of enhanced sampling methods. In hybrid QM/MM simulations, NAMD offloads part of its standard force and energy calculations to a QM program, either through native interfaces to MOPAC [106,68] or ORCA [80], or through a flexible generic interface requiring a wrapper script, where exemplary Python wrappers are provided for Gaussian, TeraChem and Q-CHEM. Multiple QM-MM coupling schemes are implemented, allowing for both mechanically and electrostatically embedded QM regions to be used (see description in Nature Methods [75]). QM/MM simulations require the same input files used for classical MD, with additional options in the configuration file. QM and MM atoms covalently bound are usually treated by redistributing the MM atom's charge over its nearest MM neighbors and by capping the QM atom with a hydrogen atom, as shown in Figure 15 for a solvated tri-alanine QM/MM calculation using the NAMD/ORCA interface. Tests of the QM/MM interface for accuracy, stability and performance, are provided as supporting information in Nature Methods [75].
If employing NAMD QM/MM please cite:
NAMD goes quantum: An integrative suite for hybrid simulations. Melo*, M. C. R.; Bernardi*, R. C.; Rudack T.; Scheurer, M.; Riplinger, C.; Phillips, J. C.; Maia, J. D. C.; Rocha, G. D.; Ribeiro, J. V.; Stone, J. E.; Neese, F.; Schulten, K.; Luthey-Schulten, Z.; Nature Methods, 2018 (doi:10.1038/nmeth.4638)
|