The measure command supplies several algorithms for analyzing molecular structures. In the following options, selection refers to an atom selection, as returned by the atomselect command described in section 9.3.2. The optional weight must be either none, an atom selection keyword such as mass, or a list of values, one for each atom in the selection, to be used as weights. If weight is missing or is none, then all weights are taken to be 1. When an atom selection keyword is used, the weights are taken from selection1.
Known Issue: The output of hbonds cannot be considered 100% accurate if the donor and acceptor selection share a common set of atoms.
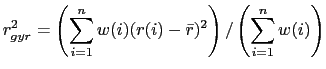 |
(9.1) |
Here are a few examples of usage:
measure bond {3 5} - Returns the distance between atoms 3 and 5 of the
current frame of the top molecule. measure bond {3 5} molid 1 frame all - Returns the distance between
atoms 3 and 5 of molecule 1 for all frames.
measure bond {3 {5 1}} molid 0 first 7 - Returns the distance between
atoms 3 of molecule 0 and atom 5 of molecule 1. The value is computed for all
frames between the seventh and the last frame of molecule 0.
Here is a 2D example of a nonorthogonal PBC cell: A and B are the are the displacement vectors which are needed to create the neighboring images. The parallelogram denotes the PBC cell with the origin O at its center. The square to the right indicates the orthonormal unit cell i.e. the area into which the atoms will be wrapped by transformation T.
+ B
/ + B'
_________/________ |
/ / / +---|---+
/ / / T | | |
/ O--------/-------> A ====> | O---|--> A'
/ / | |
/_________________/ +-------+
A = displacement vector along X-axis with length a
B = displacement vector in XY-plane with length b
A' = displacement vector along X-axis with length 1
B' = displacement vector along Y-axis with length 1
O = origin of the PBC cell
|
The second argument (cutoff) is the maximum distance (in Å) from the PBC unit cell for atoms to be considered. In other words the cutoff vector defines the region surrounding the pbc cell for which image atoms shall be constructed (i.e. {6 8 0} means 6 Å for the direction of A, 8 Å for B and no images in the C-direction).
The following options can be specified:
Options:
Result:
The return value is a TCL list of pairs consisting of a label string and a value or list. For each label the data following it are described below:
If a certain item is not present (e.g. no planes or no axes) then the corresponding value is an empty list. The pair format allows to use the result as a TCL array for convenient access of the different return items.
Example:
set sel [atomselect top all]
# Determine the symmetry
set result [measure symmetry $sel]
# Create array 'symm' containing the results
array set symm $result
# Print selected elements of the array
puts $symm(pointgroup)
puts $symm(order)
puts $symm(elements)
puts $symm(axes)
# Set atoms of selection to ideally symmetric coordinates
$sel set {x y z} $symm(ideal)
Classify the space surrounding a molecule as interior or exterior. For structures not representing a closed container, or in applications where interior and exterior boundaries are ill-defined such as surface cleft identification, fuzzy boundary detection should be enabled with the -probmap flag.
When measure volinterior is invoked, a QuickSurf density of the atom selection is first computed; the -res and -isovalue parameters correspond to QuickSurf parameters used in generating the molecular density. For each voxel not corresponding to molecular density (i.e., selection voxels), rays are cast in parallel. In the default, fixed boundary implementation: if a ray strikes the system boundary before selection voxels, then that voxel is considered exterior and is marked accordingly and ignored for the remainder of the calculation. A Tcl list is returned with four entries: total voxel count, exterior voxel count, interior voxel count, and selection voxel count. The resulting volume loaded into -mol molid is a discrete 3D grid, where each voxel is classified as interior, exterior, or selection.
For the fuzzy boundary implementation, i.e. the -probmap flag is given, all
![]() rays are cast and the number of rays striking selection voxels before the
system boundary is stored for each voxel.
The continuous grid output by the fuzzy boundary implementation is normalized
against
rays are cast and the number of rays striking selection voxels before the
system boundary is stored for each voxel.
The continuous grid output by the fuzzy boundary implementation is normalized
against ![]() rays such that all entries vary continuously on
rays such that all entries vary continuously on ![]() .
The directions of rays are randomly chosen and are uniformly distributed on the
surface of the unit sphere following a Poisson disk sampling procedure, although
uniformity of ray directions below -nrays 32 cannot be guaranteed.
Thus, values passed to -nrays of at least 32 are recommended if the
-probmap flag is given. In addition to the continuous 3D grid loaded into
-mol molid, a Tcl list is returned containing ten entries, where each
entry is the number of voxels in the grid at percentiles ranging from 10-99th.
Optionally, the -discretize flag will process the continuous grid into a
discrete grid, equivalent to that output by the fixed boundary implementation,
according to the user-specified cutoff value. If -discretize is given,
then a Tcl list of total, exterior, interior and selection voxel counts is
returned.
.
The directions of rays are randomly chosen and are uniformly distributed on the
surface of the unit sphere following a Poisson disk sampling procedure, although
uniformity of ray directions below -nrays 32 cannot be guaranteed.
Thus, values passed to -nrays of at least 32 are recommended if the
-probmap flag is given. In addition to the continuous 3D grid loaded into
-mol molid, a Tcl list is returned containing ten entries, where each
entry is the number of voxels in the grid at percentiles ranging from 10-99th.
Optionally, the -discretize flag will process the continuous grid into a
discrete grid, equivalent to that output by the fixed boundary implementation,
according to the user-specified cutoff value. If -discretize is given,
then a Tcl list of total, exterior, interior and selection voxel counts is
returned.
For more information, please refer to the publication [47]:
https://doi.org/10.1021/acs.jcim.9b00324
Options:
Fixed boundary example:
set id [mol new myMolecule.pdb]
set sel [atomselect $id "noh protein"]
set result [measure volinterior $sel -nrays 12 \
-isovalue 1.0 -spacing 1.0 -res [expr 1.2/0.2] \
-mol $id -verbose]
set totalVoxels [lindex $result 0]
set exteriorVoxels [lindex $result 1]
set interiorVoxels [lindex $result 2]
set selectionVoxels [lindex $result 3]
# Use output volume grid to make a selection
set watersInterior [atomselect $id "water and vol0 < 1 and vol0 > -1"]
Fuzzy boundary example:
set id [mol new myMolecule.pdb]
set sel [atomselect $id "noh protein"]
# Continuous grid output
set resultC [measure volinterior $sel -nrays 64 \
-isovalue 0.8 -spacing 1.0 -res [expr 1.0/0.2] \
-mol $id -verbose -probmap]
set 50th_percentile [lindex $resultC 4]
set 60th_percentile [lindex $resultC 5]
# Use output to select waters in regions above 85% likelihood of being interior
set watersInterior [atomselect $id "water and vol0 > 0.85"]
# Discrete grid output
set resultD [measure volinterior $sel -nrays 64 \
-isovalue 0.8 -spacing 1.0 -res [expr 1.0/0.2] \
-mol $id -verbose -probmap -discretize 0.85]
set totalVoxels [lindex $resultD 0]
set exteriorVoxels [lindex $resultD 1]
set interiorVoxels [lindex $resultD 2]
set selectionVoxels [lindex $resultD 3]
# Use discretized grid to select interior waters,
# equivalent to above selection on continuous grid.
## Note that we now use vol1, because we did not choose to overwrite the
## previous volume (vol0).
set watersInterior2 [atomselect $id "water and vol1 < 1 and vol1 > -1"]