



Next: menu
Up: Tcl Text Commands
Previous: material
Contents
Index
The meaure command supplies several algorithms for analyzing molecular
structures. In the following options, selection refers to an atom
selection, as returned by the atomselect command described in section
8.3.2. The optional weight must be either
none, an atom selection keyword such as mass, or a list of values,
one for each atom in the selection, to be used as weights. If weight
is missing or is none, then all weights are taken to be 1. When
an atom selection keyword is used, the weights are taken from selection1.
- center selection [weight weight]:
Returns the geometric center of atoms in selection using the
given weight.
- minmax selection:
Returns two vectors, the first containing the minimum
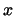 ,
, 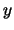 , and
, and 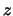 coordinates of all atoms in selection, and the second containing the
corresponding maxima.
coordinates of all atoms in selection, and the second containing the
corresponding maxima.
- rmsd selection1 selection2 [weight weight]:
Returns the root mean square distance between corresponding atoms in
the two selections, weighted by the given weight. selection1 and
selection2 must contain the same number of atoms (the selections may
be from different molecules that have different numbers of atoms).
- fit selection1 selection2 [weight weight]:
Returns a 4x4 transformation matrix which, when applied to the atoms in
selection1, minimizes the weighted RMSD between selection1 and
selection2.
- inverse matrix:
Returns the inverse of the given 4x4 matrix.
See section for more on
RMSD alignment.
Next: menu
Up: Tcl Text Commands
Previous: material
Contents
Index
vmd@ks.uiuc.edu