From: Leandro Martínez (leandromartinez98_at_gmail.com)
Date: Wed Oct 05 2016 - 15:58:39 CDT
Concerning references for RMSD calculations, you might want to take a look
at this package we developed:
http://leandro.iqm.unicamp.br/mdlovofit/
It computes the RMSD while identifying the most conserved regions within
the structure, and uses those regions as a reference for the alignment.
It can help to interpret what is actually going on in your simulations if
the
average fluctuations seem to be large.
Leandro.
On Wed, Oct 5, 2016 at 5:39 PM, Radak, Brian K <bradak_at_anl.gov> wrote:
> In my opinion, if you want to quantify the fluctuations, you have to be
> careful in what you choose as your reference for RMSD - looking for
> anomalous domains, as you have, is probably an excellent idea (and much
> more informative than simply saying "the RMSD is noisy").
>
> Long equilibration times are absolutely acceptable, especially if you are
> not so sure the initial conditions are physically realistic or relevant.
> Coming from the nucleic acids community we regularly expect equilibration
> times > 50 ns - I try to take people at their word when they say a protein
> system equilibrated faster than that, but it always seems overly fortuitous
> to me.
>
> There are a number of analyses that might be useful for comparing with B
> factors - principle component analyses is probably the most popular. A
> cluster analyses is also probably a good idea (and might provide a better
> reference for RMSD calculations).
>
> HTH,
> Brian
>
> Brian Radak
> Postdoctoral Appointee
> Leadership Computing Facility
> Argonne National Laboratory
>
> 9700 South Cass Avenue, Bldg. 240
> Argonne, IL 60439-4854
> (630) 252-8643
> brian.radak_at_anl.gov
> ------------------------------
> *From:* owner-namd-l_at_ks.uiuc.edu [owner-namd-l_at_ks.uiuc.edu] on behalf of
> Dhiraj Srivastava [dhirajks_at_gmail.com]
> *Sent:* Wednesday, October 05, 2016 2:02 PM
> *To:* namd-l_at_ks.uiuc.edu; Pardis Tabaee
> *Subject:* Re: namd-l: question regarding rmsd
>
> Thank you Everyone.
> The simulation was started from crystal structure obtained by
> cocrystallizing with ligand. I don't see any significant difference in apo
> vs ligand bound form but ligand is an inhibitor. I think for unstable
> RMSD, I can blame a small flexible domain. After removing the flexible
> domain from RMSD calculation, I got stable RMSD very similar to apo
> protein. However its still took long time to equilibrate. the sudden change
> in rmsd at around 30 ns is still there. can I use data after 40 ns for the
> analysis? is it acceptable to have such a long equilibration time? I am
> primarily a crystallographer and I don't have much experience in publishing
> articles with MD simulation data. So I don't know what's acceptable in the
> community? I am doing simulation to find out if allosteric behaviour of
> ligand is due to change in dynamics. I would like to compare it with B
> factor change in crystal structure.
>
> Thanks
> Dhiraj
>
> On Wed, Oct 5, 2016 at 1:34 PM, Pardis Tabaee <
> pardis.tabaee.d_at_hotmail.co.uk> wrote:
>
>> I think it's the average RMSD that was calculated. Maybe you can try to
>> equilibrate without the solvent and see if you reach a stable conformation
>> of your complex quicker than if you do it with the solvent? There are also
>> other strategies. SMD is one of them, where you are applying constraints,
>> you can do this with NAMD.
>> ------------------------------
>> *From:* owner-namd-l_at_ks.uiuc.edu <owner-namd-l_at_ks.uiuc.edu> on behalf of
>> Radak, Brian K <bradak_at_anl.gov>
>> *Sent:* 05 October 2016 17:18
>> *To:* namd-l_at_ks.uiuc.edu; Roshan Shrestha; dhirajks_at_gmail.com
>> *Subject:* RE: namd-l: question regarding rmsd
>>
>> I'm not clear what the "problem" is here. Are you not happy with the
>> (large?) oscillations after 100 ns? I don't understand why you would be
>> surprised that convergence to a stable state takes that long. I'm more
>> surprised that the apo state levels out in 20 ns.
>>
>> I presume this is RMSD with respect to the initial frame, as is default
>> for VMD? That's a completely arbitrary reference and thus I wouldn't hold
>> too much stock in it. Is the initial bound state from a crystal structure
>> or was it generated by docking to the apo structure? If the latter, then
>> there isn't really a reason to believe the initial configuration had a high
>> Boltzmann weight anyway - slow equilibration is to be expected and could
>> involve large conformational changes.
>>
>> Brian
>>
>> Brian Radak
>> Postdoctoral Appointee
>> Leadership Computing Facility
>> Argonne National Laboratory
>>
>> 9700 South Cass Avenue, Bldg. 240
>> Argonne, IL 60439-4854
>> (630) 252-8643
>> brian.radak_at_anl.gov
>> ------------------------------
>> *From:* owner-namd-l_at_ks.uiuc.edu [owner-namd-l_at_ks.uiuc.edu] on behalf of
>> Roshan Shrestha [roshanpra_at_gmail.com]
>> *Sent:* Wednesday, October 05, 2016 11:07 AM
>> *To:* namd-l_at_ks.uiuc.edu; dhirajks_at_gmail.com
>> *Subject:* Re: namd-l: question regarding rmsd
>>
>> Don't worry dhiraj, it's obviously rmsd in Y-axis and Time steps in
>> X-axis. Shouldn't have been any fuss with it !!!
>>
>> On Wed, Oct 5, 2016 at 9:48 PM, <dhirajks_at_gmail.com> wrote:
>>
>>> Sorry. I should have labeled it. Y axis is time in picosecond and x axis
>>> is rmsd in Angstrom.
>>>
>>> Dhiraj
>>>
>>> Sent from my iPhone
>>>
>>> On Oct 5, 2016, at 10:55 AM, Pardis Tabaee <
>>> pardis.tabaee.d_at_hotmail.co.uk> wrote:
>>>
>>> Hi,
>>>
>>>
>>> What's on the y axis?
>>>
>>>
>>> Regards,
>>>
>>>
>>> P
>>>
>>>
>>> ------------------------------
>>> *From:* owner-namd-l_at_ks.uiuc.edu <owner-namd-l_at_ks.uiuc.edu> on behalf
>>> of Dhiraj Srivastava <dhirajks_at_gmail.com>
>>> *Sent:* 04 October 2016 22:07
>>> *To:* namd-l_at_ks.uiuc.edu
>>> *Subject:* namd-l: question regarding rmsd
>>>
>>> Hi
>>> I am trying to do MD simulation on a protein with and without ligand.
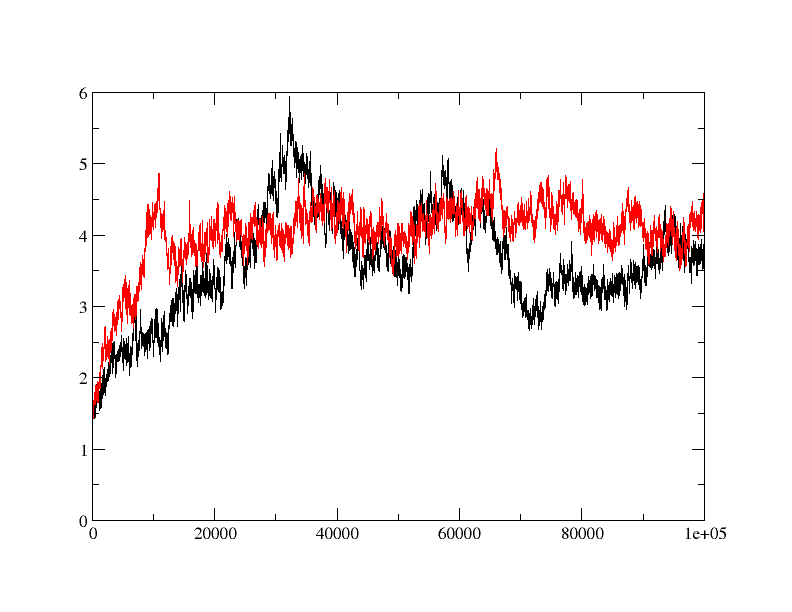
>>> when I did rmsd plot, I found that apo protein is behaving fine (red) but
>>> ligand bound form (black) is taking relatively longer time to equilibrate
>>> and showing quite a bit of fluctuation in rmsd. is the fluctuation in rmsd
>>> value for ligand bound protein is acceptable or is there anything wrong?
>>> How can I fix it?
>>>
>>> thanks
>>> Dhiraj
>>>
>>>
>>> [image: Inline image 3]
>>>
>>>
>>
>>
>> --
>> Roshan Shrestha
>> Graduate Student
>> Central Department of Physics,Tribhuvan University
>> Kathmandu,Nepal
>>
>>
>>
>
This archive was generated by hypermail 2.1.6 : Sun Dec 31 2017 - 23:20:45 CST