From: Souvik Sinha (souvik.sinha893_at_gmail.com)
Date: Tue Dec 08 2015 - 01:42:34 CST
I think increasing the sampling time will eventually decrease the barrier
until convergence of sampling is achieved . And you can also use the
"historyFreq " option to your restarin1.in file so that you can have the
track for the pmf during the simulation.
On Tue, Dec 8, 2015 at 11:18 AM, sunyzero <sunyzero_at_163.com> wrote:
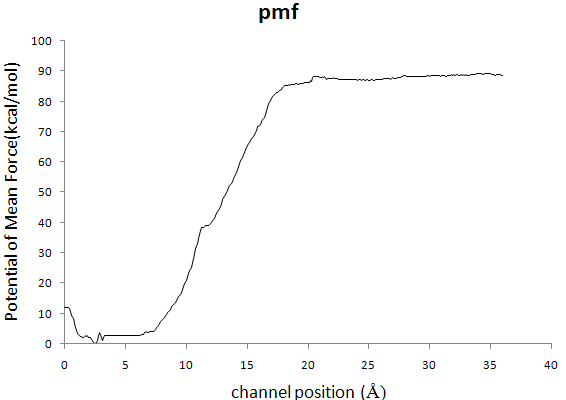
> Dear Namd users
> Hello, I need your help about determining the PMF with Adaptive Biasing
> Forces. According to tutorial about Methods for calculating Poteintials of
> Mean Force, I use it on HIV-1 protease inhibitors dissociation process,and
> inhibitors dissociate from binding site moving range from 0 to 36Å, which
> is divided into six window,each 6-Å wide,but in the ABF output .pmf file,
> the value of pmf is too large,the maximum value is 88kcal/mol. I try many
> times, there still exist questions.Here is one of my configures.
> # Force-Field Parameters
> exclude scaled1-4
> 1-4scaling 1.0
> cutoff 12.
> switching on
> switchdist 10.
> pairlistdist 13.5
> # Integrator Parameters
> timestep 1.0 ;# 2fs/step (only if needed to finish quickly)
> rigidBonds water ;# needed for 2fs steps
> nonbondedFreq 2
> fullElectFrequency 4
> stepspercycle 20
> # Constant Temperature Control
> langevin on ;# do langevin dynamicsgmail.google.com
> langevinDamping 1 ;# damping coefficient (gamma) of 5/ps
> langevinTemp $temperature
> langevinHydrogen off ;# don't couple langevin bath to hydrogens
> wrapAll on
> # PME (for full-system periodic electrostatics)
> PME yes
> PMEGridSpacing 1.0
> # Constant Pressure Control (variable volume)
> useGroupPressure no ;# needed for rigidBonds
> useFlexibleCell no
> useConstantArea no
>
> langevinPiston on
> langevinPistonTarget 1.01325 ;# in bar -> 1 atm
> langevinPistonPeriod 100.
> langevinPistonDecay 50.
> langevinPistonTemp $temperature
> # Output
> outputName win1
> restartfreq 500 ;# 1000steps = every 1ps
> dcdfreq 500
> xstFreq 500
> outputEnergies 500
> outputPressure 500
> colvars on
> colvarsConfig restrain1.in
> # Minimization
> #minimize 500
> #reinitvels $temperature
> run 1500000 ;#
>
>
> And here is my restrain1.in
> colvarsTrajFrequency 500
> colvarsRestartFrequency 500
> colvar {
> name Translocation
>
> width 0.1
>
> lowerboundary 0.0
> upperboundary 6.0
>
> lowerwallconstant 100.0
> upperwallconstant 100.0
>
> distanceZ {
> main {
> atomnumbers { the atoms of ligand }
> }
> ref {
> atomnumbers { the atoms of protein and name CA
> }
> }
> axis ( 0.0, 0.0, 1.0 )
> }
> }
>
> abf {
> colvars Translocation
> fullSamples 1000
>
>
>
>
>
-- Souvik Sinha Research Fellow Bioinformatics Centre (SGD LAB) Bose Institute Contact: 033 25693275
This archive was generated by hypermail 2.1.6 : Thu Dec 31 2015 - 23:22:18 CST