From: Jonasson Gabriella (gabriella.jonasson_at_gmail.com)
Date: Mon Jan 05 2015 - 09:44:31 CST
Dear NAMD users,
I'm trying to estimate the free energy of binding of a small molecule to a protein by means of FEP simulations. I'm using NAMD 2.9 and the parameters proposed in the tutorial "Protein:ligand standard binding free energies" by Gumbart, Roux and Chipot.
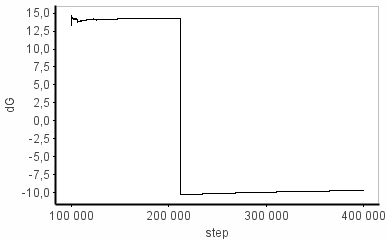
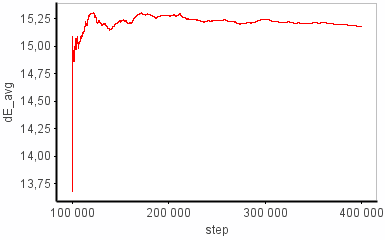
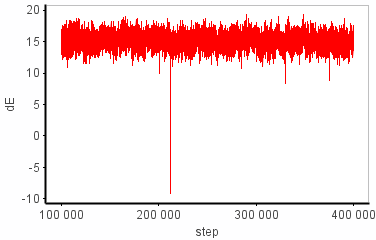
I have problems with the dG sampling in some of my simulation windows. Sometimes I see a huge change in dE from one step to the next (e.g. +14 -> -9 kcal/mol). I can't find the structural origin of this energy change (some sort of clash maybe?) because the coordinates aren't printed out as often as the FEP-energies. Anyhow, the energy difference between the two states goes back to "normal" (i.e. ~14 kcal/mol) pretty quickly (~400 fs) and this episode doesn't seem to affect dE_avg much at all. On the other hand, dG is completely put off track, where it stays for the rest of the simulation. I’ve attached the graphs that illustrate this.
I know that dG is based on an ensemble average but I don’t understand why it doesn’t behave more similarly to dE_avg under these conditions. Can anybody explain?
Is this kind of simulation useless or can I post-treat the energies somehow?
Best regards,
Gabriella
This archive was generated by hypermail 2.1.6 : Thu Dec 31 2015 - 23:21:31 CST