From: Norman Geist (norman.geist_at_uni-greifswald.de)
Date: Thu Jun 19 2014 - 02:46:48 CDT
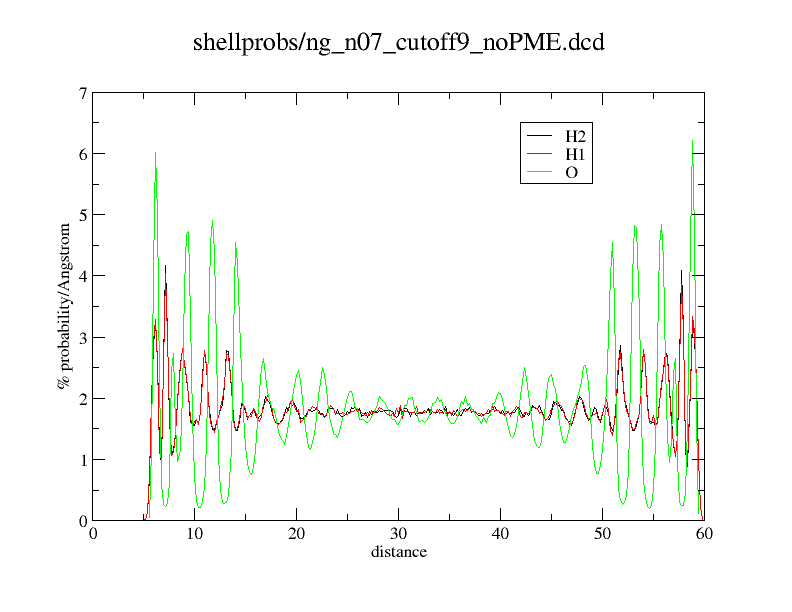
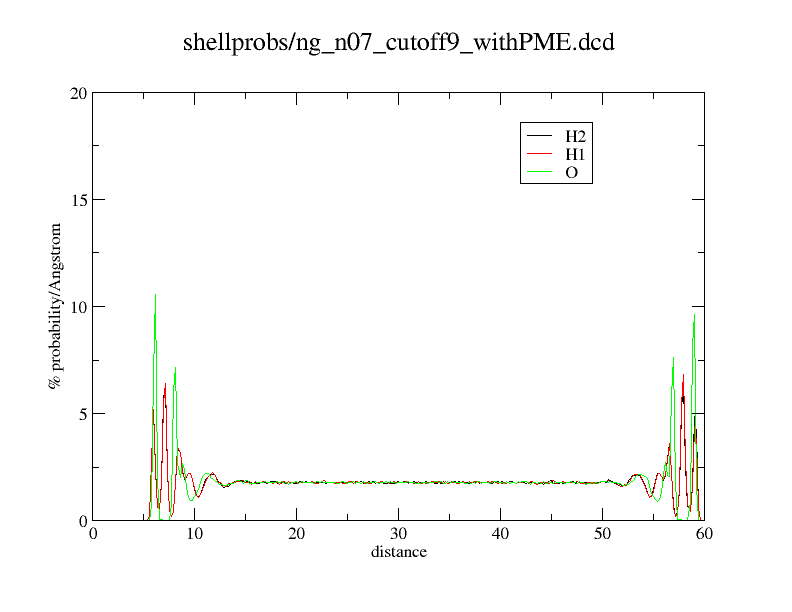
Here an example what happens. What you see is just a distance histogram of water molecules to an titanium dioxide surface.
Norman Geist.
> -----Ursprüngliche Nachricht-----
> Von: owner-namd-l_at_ks.uiuc.edu [mailto:owner-namd-l_at_ks.uiuc.edu] Im
> Auftrag von Fotis Baltoumas
> Gesendet: Mittwoch, 18. Juni 2014 19:59
> An: NAMD mailing list
> Betreff: namd-l: Should PME be used for electrostatic interaction
> energy calculations?
>
> Dear NAMD users,
>
> Having performed a number of MD simulations on protein-protein
> complexes
> with NAMD, I am currently attempting an evaluation of interaction
> energy
> values with VMD's NAMD Energy plugin. In my simulations I defined
> periodic
> boundary conditions and used the Particle Mesh Ewald method for full
> electrostatics.
> My question is: should I also use PME in the calculation of
> electrostatic
> interaction energy, or should I use the default settings, and calculate
> with Coulomb's law and the defined cutoff? I have attempted both and,
> although for some complexes the results are similar, in other cases the
> changes are dramatic. For one particular example the results are:
> Electrostatics without PME: -26.22 kcal/mol (Std. Dev.=10.16)
> Electrostatics with PME: -204.58 kcal/mol (Std. Dev.=10.53)
> In your opinion, which value should I trust as "biologically relevant"?
> Can the PME result be interpreted as the influence of distant charged
> groups to my complex, or should I use the non-PME result instead?
>
> Thank you,
> Fotis Baltoumas
--- Diese E-Mail ist frei von Viren und Malware, denn der avast! Antivirus Schutz ist aktiv. http://www.avast.com
This archive was generated by hypermail 2.1.6 : Thu Dec 31 2015 - 23:20:53 CST