From: Aron Broom (broomsday_at_gmail.com)
Date: Mon Feb 10 2014 - 11:20:54 CST
I think ABF will not sample evenly across the reaction coordinate until the
energy landscape has been flattened, so if the counts aren't even then this
suggests it hasn't reached equilibrium. In particular, it looks like you
are not sampling the energy minima well, which suggests maybe the
fullsSamples value could be increased so that the bias is not applied
prematurely. Keep in mind that the fullSamples, I think, it the number of
timesteps. So assuming a 2 fs timestep, you are talking about 80 ps of
simulation until you apply the bias. For systems such as proteins or
nucleic acids, the decorrelation time of the coordinates can easily be on
this same order, so while you have measured 40000 samples, you may in
practice only have 1 or 2 unique ones, and so the bias that gets applied
can be very far from what it "ought" to be, and then it can be a very long
road to correcting that initial mistake.
Umbrella sampling obviously avoids the above issue, since you can choose
where to invest more simulation time if you want.
On Sat, Feb 8, 2014 at 4:38 PM, Abir Ganguly <7.someone.iitd_at_gmail.com>wrote:
> Dear NAMD users,
>
> I have been trying to calculate the PMF along a particular angle
> coordinate in a relatively small RNA system. The angle is a heavy
> atom-hydrogen-heavy atom angle, and spans from ~50 to ~180 degrees. I am
> trying to do it in two different ways, using ABF simulations and umbrella
> sampling simulations.
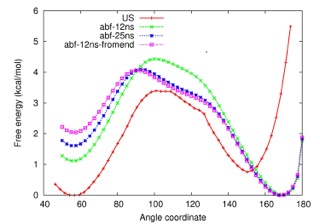
> In the case of ABF, I first ran for 12ns using fullsamples 40,000, and
> then extended the simulation to 25ns. I also ran another 12ns simulation
> starting from the end of the 25ns simulation.
> In the case of US, I manually ran 42 windows each for over 1 ns ( 200 ps
> equil). Attached are the pmf plots obtained from the simulations. As you
> can see from the plot, the results from the two methods are not consistent
> with each other. When I look at the .count file from the ABF
> simulations(plot attached), I notice that the end points are not sampled
> well. The qualitative similar behaviour between the 12ns and 25ns
> simulations to me is a bit disconcerting. Just increasing the simulation
> time may not fix the problem. I will appreciate any suggestion on how to
> improve the sampling at the end points.
>
> Below is an example colvar file that I am using for the ABF simulations
>
> ************************************************************
> ********************************
>
> colvarsTrajFrequency 100
> colvarsRestartFrequency 1000
> colvarsTrajAppend on
>
> colvar {
> name orient
> width 2.5
> lowerboundary 50
> upperboundary 180
> lowerWallConstant 1
> upperWallConstant 1
> angle {
> group1 {
> atomnumbers 29
> }
> group2 {
> atomnumbers 30
> }
> group3 {
> atomnumbers 34
> }
> }
> }
>
>
> abf {
> colvars orient
> fullSamples 40000
> inputPrefix pmf-output
> }
> ************************************************************
> ******************
>
> Thanks for your time.
>
> Best,
>
> Abir
>
>
-- Aron Broom M.Sc PhD Student Department of Chemistry University of Waterloo
This archive was generated by hypermail 2.1.6 : Thu Dec 31 2015 - 23:20:28 CST
{kind=link}
{kind=link}