From: Aron Broom (broomsday_at_gmail.com)
Date: Tue Jun 19 2012 - 16:56:51 CDT
Personally I wouldn't start from the state you want, as you might fail to
learn something interesting by doing this, for instance, that the geometry
you imagine is the best, actually isn't. I realize that it makes the
process simpler, but keep in mind that ABF is an equilibrium method, so if
you don't get to the point where your system is freely moving back and
forth between the fully unbound (stuck to nano-tube) and fully bound
(base-paired) states, then you haven't simulated for long enough anyway.
For ABF, if you haven't set the "fullsamples" variable high enough, it
might take VERY long for that to happen, and you'd be better suited to go
back and increase that variable by a factor of 10 or so. For that reason,
I would suggest that when you are initially working out the details of how
you want to bias, you should use MetaDynamics instead, although it has many
of it's own unique problems and ABF might be better in some cases, I've
found Metadynamics superior for initially getting a feel for the problem
(my opinion).
You mention that there are many degrees of freedom and that sampling over
these is part of the issue, which I agree is probably what is happening.
What you need is either a method for increasing your amount of sampling,
such that you can accurately average across the real ensemble of
configurations, or, a method that allows you to bias along the important
and slow degrees of freedom that your current colvar doesn't affect. I
would recommend 3 possible options:
1) Use Metadynamics with multiple replicas. A single metadynamics replica
will essentially give you the same result as ABF, but by having multiple
replicas you will obtain better convergence over unaccounted for degrees of
freedom (multiple replica metadynamics is implemented in NAMD, I don't
think ABF is, but maybe I'm missing something obvious there). Of course,
this is really just the same as running the normal single replica for a
very long time and averaging over the latter parts of the simulation, but
this is computationally more efficient if you have lots of hardware
available as scaling a single job across many processors is always slower
than running many jobs on a smaller set of processors.
2) Bias along more than one reaction coordinate. You can do this in ABF
since you are familiar with that, but multidimensional biasing is simpler
with Metadynamics (especially beyond 2D). In your case, you could keep
your distance variable you already have defined, and then add another
colvar, that distinguishes between your ideal geometry and the non-ideal
one, probably this will be some kind of angle or orientation, or maybe
another distance between two other important atoms.
The second option is particularly nice because in addition to getting a
more accurate answer (at least as far as the forcefield allows) you'll be
able to see the relative energetic differences between the ideal and
non-ideal geometry, and any low energy pathways between them, which might
well be quite interesting data in of itself.
The problem with the second option is that the time required to sample
across the colvars increases exponentially as you add more colvars, but you
can partially get around this by increasing the width of the bins for the
second colvar to only capture to the resolution you really need (that is,
you might have 20 distance bins for your PMF, but only need 10 angle bins).
Also, you can combine 1) and 2) to do multiple replica multiple dimension
metadynamics. I've had some recent success with this using 3 colvars and
as many as 16-48 replicas. Obviously that is starting to become
computationally prohibitive, but it depends on what is available to you.
3) Do some kind of replica exchange. I think the standard temperature
replica exchange would seem to suit itself well to your problem.
Essentially this will act to lower the energy barrier between your ideal
and non-ideal geometries such that you can properly sample the average
between them in a reasonable time-frame.
Also, you can combine option 3) with 1) and/or 2). For instance, you could
just use your current ABF colvar definition with metadynamics, but have 4
different temperatures that you are doing replica exchange between. I
believe this can be "easily" implemented by modifying the replica exchange
Umbrella sampling script. There might be an earlier message in the list
concerning this. If you are looking for this method in the literature, I
believe it is sometimes called replica exchange metadynamics, and sometimes
parallel tempering metadynamics.
You can also do bias exchange with metadynamics (similar in spirit to the
second option, only that the computational time increases only linearly,
but you also only get a 1D answer rather than the nice 2D one). Again this
I believe can be setup based on the replica exchange umbrella sampling
script that comes with NAMD 2.9.
I would recommend searching for a 2008 Metadynamics review with Parrinello
as one of the authors, it might give you some good insight into what the
problems are, as the issue you are describing is quite common (although it
does sound like you haven't run for long enough also, as you should never
get "stuck" in an ABF or metadynamics run, as the end-point should be one
in which you diffuse freely along your reaction coordinate, and in the
worst case scenario only get stuck transiently).
~Aron
On Tue, Jun 19, 2012 at 4:43 PM, Robert Johnson
<robertjo_at_physics.upenn.edu>wrote:
> Hello All,
>
> I'm interested in determining how two complementary DNA strands can
> hybridize when they are both adsorbed to a carbon nanotube.
>
> I have already performed some ABF calculations to estimate the PMF for
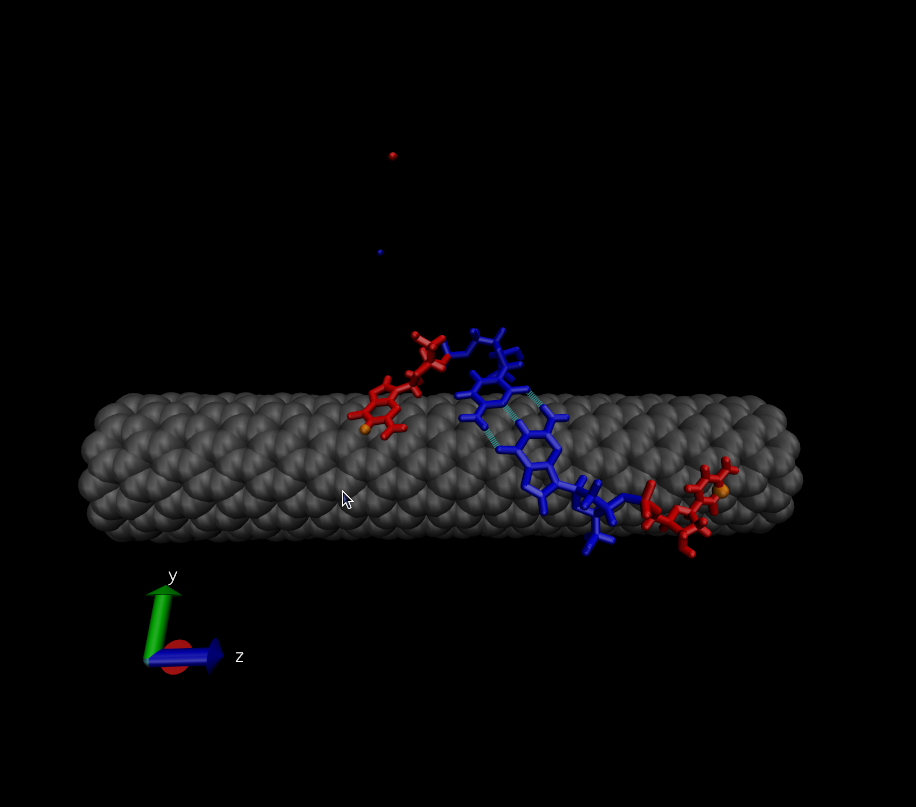
> hybridization. My initial state is shown here:
> http://www.physics.upenn.edu/~robertjo/temp/InitialState.png
>
> My system consists of 2 DNA strands that are each 2 bases long - in this
> case each strand is GC. The blue bases are forming a G-C base pair. Over
> the course of the simulation I constrain the distances between the H-bond
> donors and acceptors for this base pair. Therefore, the blue base pair is
> present throughout the entire simulation.
>
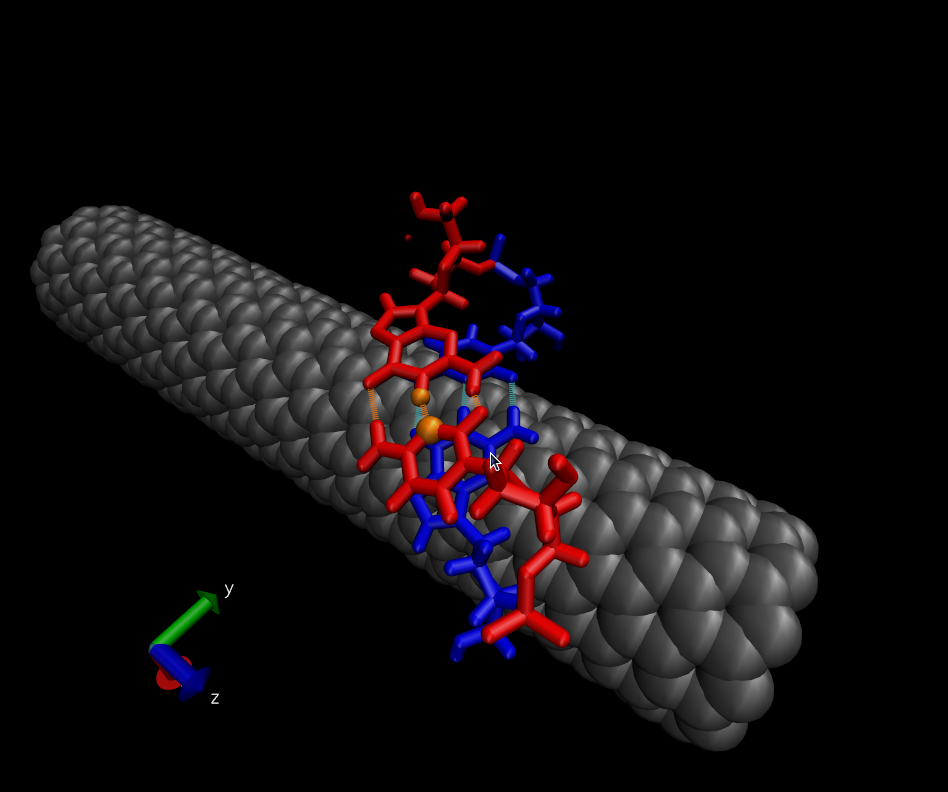
> Then ABF is employed to force the two red bases to come together. The
> collective variable used is the distance between two atoms that share a
> H-bond when the red bases are paired (the orange atoms). Applying ABF
> causes (in most cases) the red bases to move toward each other and to form
> a base pair. The only way the red bases can hybridize is by lifting off the
> surface of the nanotube. The final state is is shown here:
> http://www.physics.upenn.edu/~robertjo/temp/Hybridized.png
>
> A graph of a representative PMF of this process is shown here:
> http://www.physics.upenn.edu/~robertjo/temp/RepresentativePMF.jpg
>
> The 2 strands initially start off in a deep energy minimum corresponding
> to adsorption to the nanotube. Forcing the two red bases to hybridize
> requires the system to surmount a large energy barrier. Then the system
> falls into a small energy minimum as the bases hybridize.
>
> About 60% of the time, I obtain a similar structure (and PMF) to that
> shown in the image(s). However, the rest of the time the bases come
> together in an orientation that does not favor hybridization. This makes it
> a little bit difficult to analyze the results since it is not known ahead
> of time what pathway the molecules will take.
>
> DNA is very flexible and I doubt that I will be able to fully sample all
> the different pathways that the DNA takes to reach the hybridized state.
> However, I would like a more reliable method for forcing the system to
> reach this hybridized state.
>
> Does anyone have ideas for better collective variables to use? Would a
> different method (i.e. metadynamics or steered MD) be a better choice?
> Since I'm interested in a very specific final state, I've also considered
> starting the simulation from the hybridized state and forcing the strands
> apart.
>
> I would appreciate any feedback you could give. Thanks!
> Bob
>
> --
> Bob Johnson, PhD
> Lab Coordinator & Lecturer
> Department of Physics and Astronomy
> University of Pennsylvania
> 209 S. 33rd St.
> Philadelphia, PA 19104
> Office: David Rittenhouse Laboratory 2C11
> Phone: 215-898-5111
> http://www.physics.upenn.edu/~robertjo
>
-- Aron Broom M.Sc PhD Student Department of Chemistry University of Waterloo
This archive was generated by hypermail 2.1.6 : Tue Dec 31 2013 - 23:22:09 CST
{kind=link}
{kind=link}
{kind=link}