



Next: Investigating
Structural Alignment
Up: Phylogenetic Tree
Previous: Phylogenetic
Tree Contents
The Phylogenetic Tree feature in MultiSeq helps in determining
the
structure-based relationships between the four AARSs. To do this, it
uses a modification of Q that accounts for both
gapped and aligned regions. This new metric, QH, creates a
structure-based phylogeny that is congruent to
the sequence-based phylogenies of AARSs previously reported by Woese et
al.3
(also see Ref. 1).
To utilize this feature:
- 1 Go to Tools
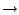 Phylogenetic Tree.
A new window entitled Phylogenetic Tree will open.
Phylogenetic Tree.
A new window entitled Phylogenetic Tree will open.
- 2 To display the structure-based Phylogenetic Tree with
appropriate labelling, see the figure.
- 3 To compute and display the tree, select by which measure
- RMSD or QH - you would like to view the tree.
For our first example, let's select all four options at the bottom of
the window and use  as our measure to generate the structure-based
relationships.
as our measure to generate the structure-based
relationships.
Can you guess as to how the protein molecules will fall relatively to
one another on the tree, considering where they fall on the
Phylogenetic tree?
Figure 20:
Example of structure-based phylogenetic tree.
|
If you guessed that the SerRS would be the furthest out and that the
P.kodakaraensis AspRS and S.cerevisiae AspRS would be
the closest, you are correct. Note that P.kodakaraensis is from
the Archaea branch of the phylogenetic tree and S.Cerevisiae is
from the Eucarya. The SerRS is used as an outlier case for this
alignment, as demonstrated in Residue Selection.
To continue to the next section of the tutorial, close the Phylogenetic
Tree window.
Next: Investigating
Structural Alignment
Up: Phylogenetic Tree
Previous: Phylogenetic
Tree Contents
Brijeet Dhaliwal
2004-09-15
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
...tein and the size of the insertion in sequence and structure.}
\end{minipage} }](img37.png)

