A CHARMM forcefield parameter file contains all of the numerical constants needed to evaluate forces and energies, given a PSF structure file and atomic coordinates. The parameter file is closely tied to the topology file that was used to generate the PSF file, and the two are typically distributed together and given matching names.
The current versions of the CHARMM forcefield are CHARMM22 for proteins and
CHARMM27 for lipids and nucleic acids including CMAP correction to proteins.
The individual parameter files are named, respectively,
par_all22_prot_cmap.inp, par_all27_lipid.prm, and
par_all27_na.prm.
To enable hybrid systems, combinations are also provided, named
par_all27_na_lipid.prm, par_all27_prot_lipid.prm, and
par_all27_prot_na.prm which can all be found in the CHARMM31 release.
While the tools used with NAMD allow multiple topology and parameter files
to be used simultaneously, it is preferable to use these pre-combined files.
The CHARMM parameters are available for download from the MacKerell web site:
We will examine par_all27_prot_lipid.prm; the other files are similar.
At the beginning of the file is the header, indicated by lines beginning
with *s:
*>CHARMM22 All-Hydrogen Parameter File for Proteins and Lipids << *>>>>> Includes phi, psi cross term map (CMAP) correction <<<<<<< *>>>>>>>>>>>>>>>>>>>>>> July, 2003 <<<<<<<<<<<<<<<<<<<<<<<<<< * All comments to ADM jr. via the CHARMM web site: www.charmm.org * parameter set discussion forum *
Comments in the parameter file are indicated by ! anywhere on a line. The next part of the file is a long set of comments containing references to the papers and other sources of the parameters in the file:
! references ! !PROTEINS ! !MacKerell, A.D., Jr,. Feig, M., Brooks, C.L., III, Extending the !treatment of backbone energetics in protein force fields: limitations !of gas-phase quantum mechanics in reproducing protein conformational !distributions in molecular dynamics simulations, Journal of !Computational Chemistry, 25: 1400-1415, 2004. ! !MacKerell, Jr., A. D.; Bashford, D.; Bellott, M.; Dunbrack Jr., R.L.; !Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; !Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F.T.K.; Mattos, !C.; Michnick, S.; Ngo, T.; Nguyen, D.T.; Prodhom, B.; Reiher, III, !W.E.; Roux, B.; Schlenkrich, M.; Smith, J.C.; Stote, R.; Straub, J.; !Watanabe, M.; Wiorkiewicz-Kuczera, J.; Yin, D.; Karplus, M. All-atom !empirical potential for molecular modeling and dynamics Studies of !proteins. Journal of Physical Chemistry B, 1998, 102, 3586-3616. !
The first set of entries in the parameter file are those for bonds,
indicated by the BONDS keyword.
Each entry consists of a pair of atom types, a spring constant, and
an equilibrium length.
The bond potential function is ![]() , where
, where ![]() is the bond length
in Angstroms.
Bonds are a stiff degree of freedom in biomolecules, so the energy function
is only accurate for values near the equilibrium length.
Entries are present for every type of bond present in the topology file.
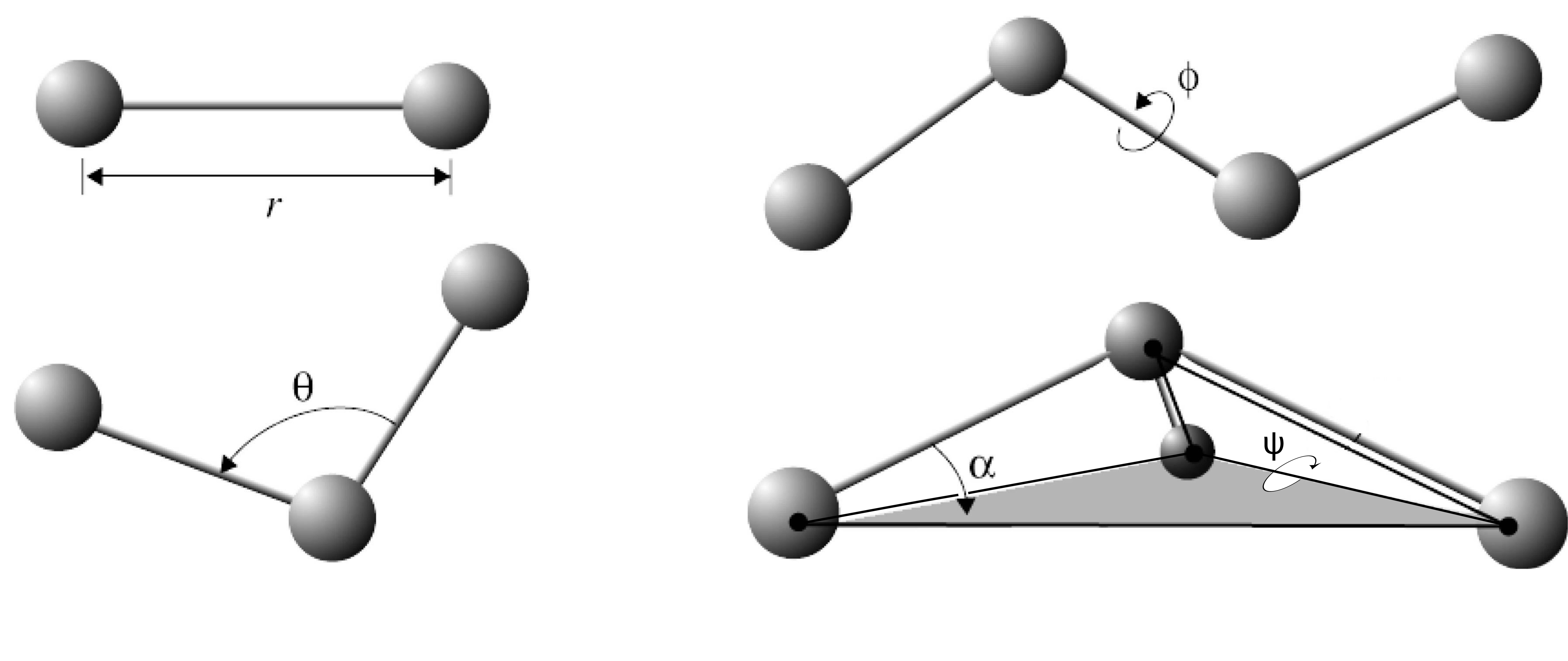
Fig. 24 illustrates how bond length, bond angle, dihedral angle and improper angle are defined.
is the bond length
in Angstroms.
Bonds are a stiff degree of freedom in biomolecules, so the energy function
is only accurate for values near the equilibrium length.
Entries are present for every type of bond present in the topology file.
Fig. 24 illustrates how bond length, bond angle, dihedral angle and improper angle are defined.
|
BONDS
!
!V(bond) = Kb(b - b0)**2
!
!Kb: kcal/mole/A**2
!b0: A
!
!atom type Kb b0
!
!Carbon Dioxide
CST OST 937.96 1.1600 ! JES
!Heme to Sulfate (PSUL) link
SS FE 250.0 2.3200 !force constant a guess
!equilbrium bond length optimized to reproduce
!CSD survey values of
!2.341pm0.01 (mean, standard error)
!adm jr., 7/01
C C 600.000 1.3350 ! ALLOW ARO HEM
! Heme vinyl substituent (KK, from propene (JCS))
CA CA 305.000 1.3750 ! ALLOW ARO
! benzene, JES 8/25/89
CE1 CE1 440.000 1.3400 !
! for butene; from propene, yin/adm jr., 12/95
CE1 CE2 500.000 1.3420 !
! for propene, yin/adm jr., 12/95
CE1 CT2 365.000 1.5020 !
! for butene; from propene, yin/adm jr., 12/95
CE1 CT3 383.000 1.5040 !
! for butene, yin/adm jr., 12/95
CE2 CE2 510.000 1.3300 !
! for ethene, yin/adm jr., 12/95
The CHARMM potential function is designed to confuse physicists, as the
form is ![]() , rather than the traditional
, rather than the traditional
![]() , and therefore the
, and therefore the ![]() given in the parameter
files is half the value of a traditional spring constant.
Also, while the units kcal/mol for energy, Angstroms for length, atomic masses,
electron charges, and either fs or ps for time may be convenient for input
and output, tortuous unit conversions are required to express the equations
of motion.
given in the parameter
files is half the value of a traditional spring constant.
Also, while the units kcal/mol for energy, Angstroms for length, atomic masses,
electron charges, and either fs or ps for time may be convenient for input
and output, tortuous unit conversions are required to express the equations
of motion.
The next section gives parameters for every type of angle present in the
topology file, indicated by the ANGLES keyword.
The angle potential function is
![]() , where
, where ![]() is the measure of the angle
in degrees.
Angles are a stiff degree of freedom in biomolecules as well, so the energy
function is only accurate for values near the equilibrium angle.
Since angles are formed from combinations of bonds, there are many more
types of angles than types of bonds.
Each entry consists of three atom types, a spring constant, and an
equilibrium angle.
A small minority of entries also contant Urey-Bradley parameters, which
are a spring constant and equilibrium length for a bond-like term between
the first and third atoms in the angle.
The beginning of the angles section is shown below, with a Urey-Bradley
term in the first entry only:
is the measure of the angle
in degrees.
Angles are a stiff degree of freedom in biomolecules as well, so the energy
function is only accurate for values near the equilibrium angle.
Since angles are formed from combinations of bonds, there are many more
types of angles than types of bonds.
Each entry consists of three atom types, a spring constant, and an
equilibrium angle.
A small minority of entries also contant Urey-Bradley parameters, which
are a spring constant and equilibrium length for a bond-like term between
the first and third atoms in the angle.
The beginning of the angles section is shown below, with a Urey-Bradley
term in the first entry only:
ANGLES
!
!V(angle) = Ktheta(Theta - Theta0)**2
!
!V(Urey-Bradley) = Kub(S - S0)**2
!
!Ktheta: kcal/mole/rad**2
!Theta0: degrees
!Kub: kcal/mole/A**2 (Urey-Bradley)
!S0: A
!
!atom types Ktheta Theta0 Kub S0
!
!Carbon Dioxide, JES
OST CST OST 3000.00 180.0000 ! CO2, JES
!Heme to Sulfate (PSUL) link
CS SS FE 50.0 100.6 !force constant a guess
!equilibrium angle optimized to reproduce
!CSD survey values
!107.5pm0.6 (mean, standard error)
!adm jr., 7/01
SS FE NPH 100.0 90.0 !force constant a guess
!adm jr., 7/01
!
CA CA CA 40.000 120.00 35.00 2.41620 ! ALLOW ARO
! JES 8/25/89
CE1 CE1 CT2 48.00 123.50 !
! for 2-butene, yin/adm jr., 12/95
CE1 CE1 CT3 48.00 123.50 !
! for 2-butene, yin/adm jr., 12/95
CE1 CT2 CT3 32.00 112.20 !
! for 1-butene; from propene, yin/adm jr., 12/95
CE2 CE1 CT2 48.00 126.00 !
! for 1-butene; from propene, yin/adm jr., 12/95
CE2 CE1 CT3 47.00 125.20 !
! for propene, yin/adm jr., 12/95
The next section gives parameters for every type of dihedral present in
the topology file; there are even more dihedrals than there are angles.
Since dihedrals represent the energy of rotation around a covalent bond,
which is the source of most conformational flexibility in biomolecules,
they must provide a smooth energy for 360 degrees.
This is done in most cases with a single sinusoid,
![]() where
where ![]() is the
angle between the plane containing the first three atoms in the dihedral and
the plane containing the last three.
The ``multiplicity''
is the
angle between the plane containing the first three atoms in the dihedral and
the plane containing the last three.
The ``multiplicity'' ![]() is typically 1, 2, or 3,
although for a small number of cases two or three terms with different
values of
is typically 1, 2, or 3,
although for a small number of cases two or three terms with different
values of ![]() are provided for the same atom types.
You may can observe in the excerpts below that the dihedral spring
constants are one to two orders of magnitude lower than for angles, with
an order of magnitude difference between flexible and inflexible dihedrals.
are provided for the same atom types.
You may can observe in the excerpts below that the dihedral spring
constants are one to two orders of magnitude lower than for angles, with
an order of magnitude difference between flexible and inflexible dihedrals.
DIHEDRALS
!
!V(dihedral) = Kchi(1 + cos(n(chi) - delta))
!
!Kchi: kcal/mole
!n: multiplicity
!delta: degrees
!
!atom types Kchi n delta
!
!Heme to Sulfate (PSUL) link
X FE SS X 0.0000 4 0.00 ! guess
!adm jr., 7/01
X CS SS X 0.0000 3 0.20 ! guess
!from methanethiol, HS S CT3 HA
!adm jr., 7/01
C CT1 NH1 C 0.2000 1 180.00 ! ALLOW PEP
! ala dipeptide update for new C VDW Rmin, adm jr., 3/3/93c
C CT2 NH1 C 0.2000 1 180.00 ! ALLOW PEP
! ala dipeptide update for new C VDW Rmin, adm jr., 3/3/93c
C N CP1 C 0.8000 3 0.00 ! ALLOW PRO PEP
! 6-31g* AcProNH2, ProNH2, 6-31g*//3-21g AcProNHCH3 RLD 4/23/93
CA CA CA CA 3.1000 2 180.00 ! ALLOW ARO
! JES 8/25/89
CA CPT CPT CA 3.1000 2 180.00 ! ALLOW ARO
! JWK 05/14/91 fit to indole
Because of the large numbers of dihedral terms required to describe a complete protein, the wildcard atom type X is occasionally used. These parameters will be used in NAMD if a more specific match is not found elsewhere in the parameter file.
!X C C X 4.0000 2 180.00 ! ALLOW HEM
! Heme (6-liganded): substituents (KK 05/13/91)
X C NC2 X 2.2500 2 180.00 ! ALLOW PEP POL ARO
! 9.0->2.25 GUANIDINIUM (KK)
X CD OH1 X 2.0500 2 180.00 ! ALLOW PEP POL ARO ALC
! adm jr, 10/17/90, acetic acid C-Oh rotation barrier
X CD OS X 2.0500 2 180.00 ! ALLOW PEP POL
! adm jr. 3/19/92, from lipid methyl acetate
X CE1 CE1 X 0.1500 1 0.00
! 2-butene, adm jr., 2/00 update
X CE1 CE1 X 8.5000 2 180.00
! 2-butene, adm jr., 2/00 update
X CE2 CE2 X 4.9000 2 180.00 !
! for ethene, yin/adm jr., 12/95
The final bond-like terms in the parameter file are impropers, which are
used exclusively and explicitly in the molecular topology to maintain
planarity.
As such, the harmonic form
![]() with a large spring
constant and
with a large spring
constant and ![]() typically zero is used to restrain deformations
among an atom and three atoms bonded to it.
As with dihedrals,
typically zero is used to restrain deformations
among an atom and three atoms bonded to it.
As with dihedrals, ![]() is angle between the plane containing the first
three atoms and the plane containing the last three.
Notice below that wildcard atom types occur in the second and third
positions, rather than the first and fourth as in dihedrals.
is angle between the plane containing the first
three atoms and the plane containing the last three.
Notice below that wildcard atom types occur in the second and third
positions, rather than the first and fourth as in dihedrals.
IMPROPER
!
!V(improper) = Kpsi(psi - psi0)**2
!
!Kpsi: kcal/mole/rad**2
!psi0: degrees
!note that the second column of numbers (0) is ignored
!
!atom types Kpsi psi0
!
CPB CPA NPH CPA 20.8000 0 0.0000 ! ALLOW HEM
! Heme (6-liganded): porphyrin macrocycle (KK 05/13/91)
CPB X X CE1 90.0000 0 0.0000 ! ALLOW HEM
! Heme (6-liganded): substituents (KK 05/13/91)
CT2 X X CPB 90.0000 0 0.0000 ! ALLOW HEM
! Heme (6-liganded): substituents (KK 05/13/91)
CT3 X X CPB 90.0000 0 0.0000 ! ALLOW HEM
! Heme (6-liganded): substituents (KK 05/13/91)
!HA C C HA 20.0000 0 0.0000 ! ALLOW PEP POL ARO
The next section gives the interpolation values for CMAP, which is a correction map to
the backbone dihedral energy. Each ![]() and
and ![]() dihedral angle, ranging from -180 to 180, is
divided into 24 grid points; the energy correction at each point is given.
A continuous function can be constructed using these values along with an interpolation formula,
which can be found in the references below.
dihedral angle, ranging from -180 to 180, is
divided into 24 grid points; the energy correction at each point is given.
A continuous function can be constructed using these values along with an interpolation formula,
which can be found in the references below.
CMAP ! 2D grid correction data. The following surfaces are the correction ! to the CHARMM22 phi, psi alanine, proline and glycine dipeptide surfaces. ! Use of CMAP requires generation with the topology file containing the ! CMAP specifications along with version 31 or later of CHARMM. Note that ! use of "skip CMAP" yields the charmm22 energy surfaces. ! ! references !MacKerell, A.D., Jr., Feig, M., Brooks, C.L., III, Accurate Treatment of !Protein Backbone Conformational Energetics in Empirical Force Fields, Submitted !for publication. !MacKerell, A.D., Jr., Feig, M., Brooks, C.L., III, Improved Treatment of the !Protein Backbone in Empirical Force Fields, Journal of the American Chemical !Society, In Press. ! alanine map C NH1 CT1 C NH1 CT1 C NH1 24 !-180 0.126790 0.768700 0.971260 1.250970 2.121010 2.695430 2.064440 1.764790 0.755870 -0.713470 0.976130 -2.475520 -5.455650 -5.096450 -5.305850 -3.975630 -3.088580 -2.784200 -2.677120 -2.646060 -2.335350 -2.010440 -1.608040 -0.482250 !-165 -0.802290 1.377090 1.577020 1.872290 2.398990 2.461630 2.333840 1.904070 1.061460 0.518400 -0.116320 -3.575440 -5.284480 -5.160310 -4.196010 -3.276210 -2.715340 -1.806200 -1.101780 -1.210320 -1.008810 -0.637100 -1.603360 -1.776870
Finally we come to the nonbonded interaction parameters. The NONBONDED statement includes a list of parameters which are used as defaults by the program CHARMM, but are ignored by NAMD. Those shown below correspond to the NAMD settings exclude scaled1-4, switching on, pairlistdist 14.0, cutoff 12.0, switchdist 10.0, dielectric 1.0, and 1-4scaling 1.0:
NONBONDED nbxmod 5 atom cdiel shift vatom vdistance vswitch -
cutnb 14.0 ctofnb 12.0 ctonnb 10.0 eps 1.0 e14fac 1.0 wmin 1.5
!adm jr., 5/08/91, suggested cutoff scheme
Recall that the partial charge of each atom is specified in the topology
and PSF files and is independent of the atom type.
Therefore the only type-based parameters are for the van der Waals
interactions, which are represented by the classic Lennard-Jones potential
(expressed in the somewhat unconventional form)
![]() .
Observe that at
.
Observe that at ![]() the force is zero and the energy is
the force is zero and the energy is
![]() .
Rather than providing a different value of epsilon could be provided for
every possible combination of atom types, only one value is provided per
type and inter-type interactions are calculated using the sum
of the radii
.
Rather than providing a different value of epsilon could be provided for
every possible combination of atom types, only one value is provided per
type and inter-type interactions are calculated using the sum
of the radii ![]() and the geometric mean of the well-depths
and the geometric mean of the well-depths ![]() .
By convention, the
.
By convention, the ![]() values are negative in the parameter file,
as seen here:
values are negative in the parameter file,
as seen here:
!
!V(Lennard-Jones) = Eps,i,j[(Rmin,i,j/ri,j)**12 - 2(Rmin,i,j/ri,j)**6]
!
!epsilon: kcal/mole, Eps,i,j = sqrt(eps,i * eps,j)
!Rmin/2: A, Rmin,i,j = Rmin/2,i + Rmin/2,j
!
!atom ignored epsilon Rmin/2 ignored eps,1-4 Rmin/2,1-4
!
C 0.000000 -0.110000 2.000000 ! ALLOW PEP POL ARO
! NMA pure solvent, adm jr., 3/3/93
CA 0.000000 -0.070000 1.992400 ! ALLOW ARO
! benzene (JES)
CC 0.000000 -0.070000 2.000000 ! ALLOW PEP POL ARO
! adm jr. 3/3/92, acetic acid heat of solvation
CD 0.000000 -0.070000 2.000000 ! ALLOW POL
! adm jr. 3/19/92, acetate a.i. and dH of solvation
CE1 0.000000 -0.068000 2.090000 !
! for propene, yin/adm jr., 12/95
CE2 0.000000 -0.064000 2.080000 !
! for ethene, yin/adm jr., 12/95
CM 0.000000 -0.110000 2.100000 ! ALLOW HEM
! Heme (6-liganded): CO ligand carbon (KK 05/13/91)
When the scaled1-4 exclusion policy is used (as it should with the CHARMM force field) nonbonded interactions of atoms separated by three bonds (i.e., atoms 1 and 4 in the chain 1-2-3-4) are modified. Even if the scaling factor for electrostatics is 1.0 (as it should be for modern CHARMM force fields), special modified van der Waals parameters are used for 1-4 pairs of atoms for which they are specified, as in the examples below.
!atom ignored epsilon Rmin/2 ignored eps,1-4 Rmin/2,1-4
!
CP1 0.000000 -0.020000 2.275000 0.000000 -0.010000 1.900000 ! ALLOW ALI
! alkane update, adm jr., 3/2/92
CP2 0.000000 -0.055000 2.175000 0.000000 -0.010000 1.900000 ! ALLOW ALI
! alkane update, adm jr., 3/2/92
CP3 0.000000 -0.055000 2.175000 0.000000 -0.010000 1.900000 ! ALLOW ALI
! alkane update, adm jr., 3/2/92
The parameter file ends with a reference to parameters for explicit hydrogen bond energy terms. These are obsolete, no longer present in the CHARMM force field, and therefore not implemented by NAMD.
HBOND CUTHB 0.5 ! If you want to do hbond analysis (only), then use
! READ PARAM APPEND CARD
! to append hbond parameters from the file: par_hbond.inp
END
For information on how the parameters have been derived, you must consult the corresponding publications referenced in the parameter files themselves or listed on the MacKerell web site http://mackerell.umaryland.edu/charmm_ff.shtml.