da.psf -- protein structure
imd_ini.pdb -- initial coordinates
par_all27_prot_lipid.prm -- CHARMM parameters
imd.namd -- NAMD configuration
imdfixedatoms.pdb -- fixed atoms
The following part of imd.namd enables IMD (already in the file):
IMDon on IMDport 3000 ;# port number (enter it in VMD) IMDfreq 1 ;# send every 1 frame IMDwait yes ;# wait for VMD to connect before running?Run NAMD:
tbss> namd2 imd.namd >! da_imd.log &The simulation will not actually run until the connection between NAMD and VMD is established.
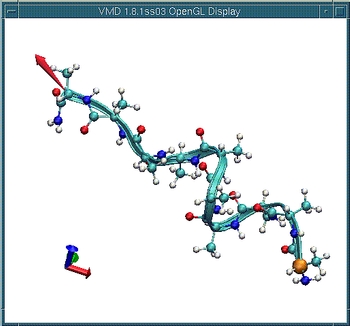
tbss> vmd -e imd.vmdThis will load the molecule and show it in both the Ribbon and the CPK representations, as saved in the VMD state file imd.vmd. You will see one atom (alpha-carbon of the first residue) colored in orange. That atom will be fixed during the IMD simulation.
Within VMD, select the menu item Extensions -> imd .
In the IMD Connection window, enter Hostname: localhost and Port: 3000
Click Connect.
You should see the molecule jiggling as the simulation is running.
Even though the double representation of Ribbon and CPK is good for viewing the overall and detailed structure of the molecule, you may change the representation to whatever you want. You can change the representation while the IMD simulation is running.
|
Now let's apply a force to stretch the molecule. Select the menu item Mouse -> Force -> Atom. Click and drag on an atom located near the other side of the `orange' atom. The red arrows indicates the force being applied (magnitude and direction). Stretch the molecule, say, by half of its original length. Now remove the force by middle-clicking on the atom to which the force is being applied. Observe how the molecule folds back.
| 
|
If you want to stop the simulation, click Stop Sim in the IMD Connection window. Otherwise, the simulation is set to run for 100 ps (about 10-20 minutes of your time). Try various things: pull different atoms, squash the molecule, do whatever you want. Use your imagination.
When the simulation stops, the molecule will stop jiggling.
Quit VMD (the menu item File -> Quit).
If you want to run the IMD simulation again, repeat the procedure of this section starting with running NAMD.
Later in the Summer School, you will be introduced to a VMD extension called AutoIMD, which is another way of doing IMD simulations. It automatically generates all the necessary files and launch and connect to a NAMD simulation.