



Next: wait
Up: Tcl Text Commands
Previous: vmdinfo
Contents
Index
volmap
The volmap command creates volumetric maps (3D grids containing a value at
each grid point) based on the molecular data, which can then be visualized in
VMD using the Isosurface and VolumeSlice representations or using the Volume
coloring mode. Also note that the VolMap plugin, accessible from the VMD
Extension menu, provides a graphical front-end to many of the volmap
command's capabilities.
To create a volumetric map, the volmap command is run in the following
way, where the atom selection specifies the atoms and molecule to include in the
calculation, and where the maptype specifies the type of volumetric data to
create:
volmap <atom selection> <maptype> [optional arguments]
For example, to create a mass density map with a cell side of 0.5 Å,
averaged over all frames of the top molecule, and add the volumetric data to the
top molecule, on would use:
volmap [atomselect top "all"] density -res 0.5 -weight mass -allframes \
-combine avg -mol top
The various volumetric data map types currently supported by volmap are
listed as follows. Please note that when a map type refer's to an atoms radius
or beta field, etc., that these values will be read directly from VMD's
associated fields for that atom. In certain cases, you may want to adjust the
atom selections fields (such as radius, beta, etc.) before performing the
volmap analysis.
- density: creates a map of the weighted atomic density at each
gridpoint. This is done by replacing each atom in the selection with a
normalized gaussian distribution of width (standard deviation) equal to its
atomic radius. The gaussian distribution for each atom is then weighted using
an optional weight (see the -weight argument), and defaults to a weight
of one (i.e, the number density). The various gaussians are then
additively distributed on a grid.
- distance: creates a map for which each gridpoint contains the
distance between that point and the edge of the nearest atom. In other words,
each gridpoint specifies the maximum radius of a sphere cnetered at that point
which does not intersect with the spheres of any other atoms. All atoms are
treated as spheres using the atoms' VMD radii.
- ligand: creates a map of the estimated potential of mean force (in
units of k
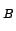 T at 300 K) of placing a weakly-interacting gas monoatomic or
diatomic ligand at every gridpoint. These results will only be valid when
averaging over a large set of frames using the -combine pmf option. Like
slowligand but uses an optimized algorithm (for example, it calculates
far-away interactions less often). Unlike slow ligand, it can also computes
many samples per gridpoint, which makes the pmf map more accurate. Please
refer to and cite: Cohen, J., A. Arkhipov, R. Braun and K. Schulten, "Imaging the
migration pathways for O
T at 300 K) of placing a weakly-interacting gas monoatomic or
diatomic ligand at every gridpoint. These results will only be valid when
averaging over a large set of frames using the -combine pmf option. Like
slowligand but uses an optimized algorithm (for example, it calculates
far-away interactions less often). Unlike slow ligand, it can also computes
many samples per gridpoint, which makes the pmf map more accurate. Please
refer to and cite: Cohen, J., A. Arkhipov, R. Braun and K. Schulten, "Imaging the
migration pathways for O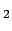 , CO, NO, and Xe inside myoglobin", 2006 (Submitted). See additional information about the ligand map type below.
, CO, NO, and Xe inside myoglobin", 2006 (Submitted). See additional information about the ligand map type below.
- slowligand: Like ligand, but uses a slower and more
rigourous algorithm. It is used to test the validity of ligand under
unusual conditions. See additional information about the ligand map type below.
- mask: creates a map which is set to 0 or 1 depending on whether
they are within a specified cutoff distance (use the -cutoff argument)
of any atoms in the selection. The mask map is typically used in combination
with other maps in order to hide/mask data that is far from a region of
interest.
- occupancy: Each grid point is set to either 0 or 1, depending on
whether it contains onbe or more atoms or not. When averaged over many frames,
this will provide the fractional occupancy of that grid point. By default,
atoms are treated as spheres using the atomic radii and a gridpoint is
considered to be "occupied" if it lies inside that sphere. Use the -points argument to treat atoms as points (a grid point is "occupied" if its
grid cube contains an atom's center).
The following optional arguments are understood by most volmap map types. Some
arguments may only apply to certain map types or may have different meaning for
different map types.
- -allframes: Use every frame in the molecule instead of just the
current one to compute the volumetric map. The method used to combine the
various trajectory frame maps can be specified using the -combine
argument. By default, volmap only uses the current frame.
- -combine < avg | max | min | stdev | pmf >: Specifies the rule to use to combine frames when using the
-allframes argument. These correspond to keeping the average, maximum or
minimum values from the range of calculated frames. stdev will return
the standard deviation for each point over the range of frames, and pmf
uses a thermal average
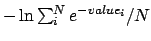 for each point. The
default is avg.
for each point. The
default is avg.
- -cutoff cutoff: Specifies a cutoff distance. For the
distance maps, specifies the largest distance that will be considered (large
number is better but slower). For the mask maps, specifies the diatnce from
each atom which will be considered part of the mask.
- -mol < molid | top >: Exports the final
volumetric data into the VMD molecule specified by molid. By default,
all maps are exported to a file or name maptype_out.dx; using the -mol option overrides this.
- -o filename: Exports the final volumetric data into a DX
file (.dx extension is added if missing). By default, all maps are exported to
a file or name maptype_out.dx.
- -points: For the occupancy map type. Treat atoms as point
particles intead of as spheres.
- -radscale factor: For the density map type. Sets a
multiplication factor that multiplies all the VMD atomic radii for the purpose
of the calculation.
- -res resolution: Sets the resolution of the map. This means
that the volume will be subdivided into many small cubes whose side have a
length of resolution.
- -weight < field name | value list >: For the
density map type. Sets a per-atom weight to be used when computing the
density. This can be the name of any VMD numerical atomic field (such as mass,
charge, beta, occupancy, user, radius, etc.) or else a Tcl list of
numbers of the same length as the number of atoms.
For the ligand and slowligand map types, the various frames should be averaged
using the ``pmf" combination rule, over very many frames. Before starting the
computation, the atomic radii of each atom should be set to the corresponding
CHARMM Lennard-Jones Rmin/2 parameter, and the beta value of each atom should
be set to the CHARMM Lennard-Jones epsilon (energy well depth) parameter. This
can be done using VMD's VolMap plugin. Simply call in succession the following
commands within the VMD console environment to use default CHARMM values for the
various atoms of a molecule:
package require volmapgui
VolMapGUI::readcharmmparams [optional list of CHARMM parameter files]
VolMapGUI::assigncharmmparams <molid>
The following optional arguments are understood only by the ligand and/or
slowligand map types.
- -dimono: Use a diatomic molecule probe with two identical atoms, instead of using a single atom as a probe.
- -dihetero: Use a diatomic molecule probe with two different atoms, instead of using a single atom as a probe.
- -conf numconf: Sample a number
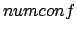 of different random rotamers of a diatomic molecule at each grid point.
of different random rotamers of a diatomic molecule at each grid point.
- -probe1 epsilon Rmin/2: Set the CHARMM van der Waals/Lennard-Jones parameters for the monoatomic probe, or for the first atom of a diatomic molecule. Units of
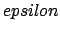 are kcal/mol, and of Rmin/2 are Å.
are kcal/mol, and of Rmin/2 are Å.
- -probe2 epsilon Rmin/2: Set the CHARMM van der Waals/Lennard-Jones parameters for the second atom of a diatomic molecule.
- -bond length: Set the bondlenth of a diatomic molecule. Units of Å.
- -cutoff cutoff: Set the CHARMM van der Waals cutoff beyong which the interaction between the probe and protein atoms is set to zero.
Next: wait
Up: Tcl Text Commands
Previous: vmdinfo
Contents
Index
vmd@ks.uiuc.edu