When one has two similar structures, one often wants to compare them. What's the difference between two X-ray structures? How much did the structure change during a simulation? To answer these questions, you must first figure out how to compare two structures, which usually means that you must find the root mean square deviation (RMSD).
Formally, given N atom positions from structure x and the
corresponding N atoms from structure y with a weighting factor
![]() ,
the RMSD is defined as:
,
the RMSD is defined as:
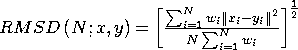
Using this equation by itself probably won't give you the answer you are looking for. Imagine two identical structures offset by some distance. The RMSD should be 0, but the offset prevents that from happening. What you really want is the minimum RMSD between two given structures; the best fit. There are many ways to do this, but for VMD we have implemented the method of Kabsch (Acta Cryst. (1978) A34, 827-828 or see file Measure.C in the VMD source code). This algorithm computes the transformation, needed to move one structure onto another in order to minimize the RMSD.
With the mathematical prerequisites behind us, we still need to be able to specify how to choose the atoms to compare. If you want to compare all the atoms in both structures, and they both have the same number of atoms, then the problem is easy - N is everything. This occurs most often in MD simulations when the only thing different between two structures are the coordinates.
But what about homologous sequences? In this case, the number of
atoms differ because while the number of residues is the same, the
sidechains have different numbers of atoms. The usual solution is to
determine the RMSD based solely on the backbone atoms or, in some
X-ray structures where only the ![]() atoms have been
determined, based on the
atoms have been
determined, based on the ![]() atoms. In addition, VMD allows two other
methods for fitting. Fitting by heavy atoms omits the hydrogens,
since their positions are often not well determined. Fitting by
``picked atoms'' performs only a translation to bring one atom directly
on top of another molecule.
atoms. In addition, VMD allows two other
methods for fitting. Fitting by heavy atoms omits the hydrogens,
since their positions are often not well determined. Fitting by
``picked atoms'' performs only a translation to bring one atom directly
on top of another molecule.
Hopefully the previous discussion revealed the importance of the options available in the fit submenu. Before examining each of them in turn, you should be aware of some VMD definitions. In VMD, a ``molecule'' refers to all the atoms from a structure file. The file may contain multiple molecules under standard chemical usage, but VMD still thinks of them as one molecule. Instead, those individual parts are called ``fragments,'' for lack of a better term. With this said, the fit options in the submenu include:
Presently the VMD feedback for RMSD picks is a little terse. To make things clear, here is the process: First select one of the options (i.e., All atoms; Heavy atoms; etc.) from the fit submenu. VMD will respond with a confirmation of your choice and then it will ask you to click on one atom of each of the two selections you would like to compare. The selection you click on first will be moved to the selection you click on second. The atoms that you click on are representative of the entire molecule, fragment, set of heavy atoms, etc. associated with those atoms. Thus, if you have chosen to RMSD fit two molecules, then clicking on one atom for each of the two molecules does in fact specify an RMSD calculation involving all atoms of the two molecules.
As a simple example of computing RMSD, load the same molecule twice. Press the `8' key (this puts VMD into the ``MoveMolecule'' pick mode and lets you use the mouse to change the coordinates of a molecule). Click and drag one of the molecules away from the other so there is a space between the two.
By default the fit routines are configured to do the best fit but for
this example you will only compute the RMSD, so pick ``Fit
![]() Print RMSD.'' Now go to the pop-up menu and pick ``Fit
Print RMSD.'' Now go to the pop-up menu and pick ``Fit
![]() Two Molecules
Two Molecules ![]() All Atoms.'' Look at the
VMD console window and you'll see a new line was printed to show the
current state. The mouse should look like a crosshair. Pick one of
the atoms in the first molecule and then one of the atoms in the second
molecule. The value of the RMSD will be printed to the screen, for
example:
All Atoms.'' Look at the
VMD console window and you'll see a new line was printed to show the
current state. The mouse should look like a crosshair. Pick one of
the atoms in the first molecule and then one of the atoms in the second
molecule. The value of the RMSD will be printed to the screen, for
example:
RMSD between the two molecules is: 1.123410
In case of two identical molecules the RMSD calculation should, of course,
give zero as a result.
To stop computing RMSDs, simply change the mouse mode to what
you want to do next, for example, press r to go to the rotate mode.
A discussion of advanced RMSD features available through the scripting interface can be found in section §.
VMD's atom alignment should be able to handle most common tasks, but
there are some it cannot do. If this occurs, you might want to look
at Andrew Martin's ProFit at
http://www.biochem.ucl.ac.uk/~martin/text/ProFit.readme