From: Giacomo Fiorin (giacomo.fiorin_at_gmail.com)
Date: Thu Oct 08 2020 - 09:36:35 CDT
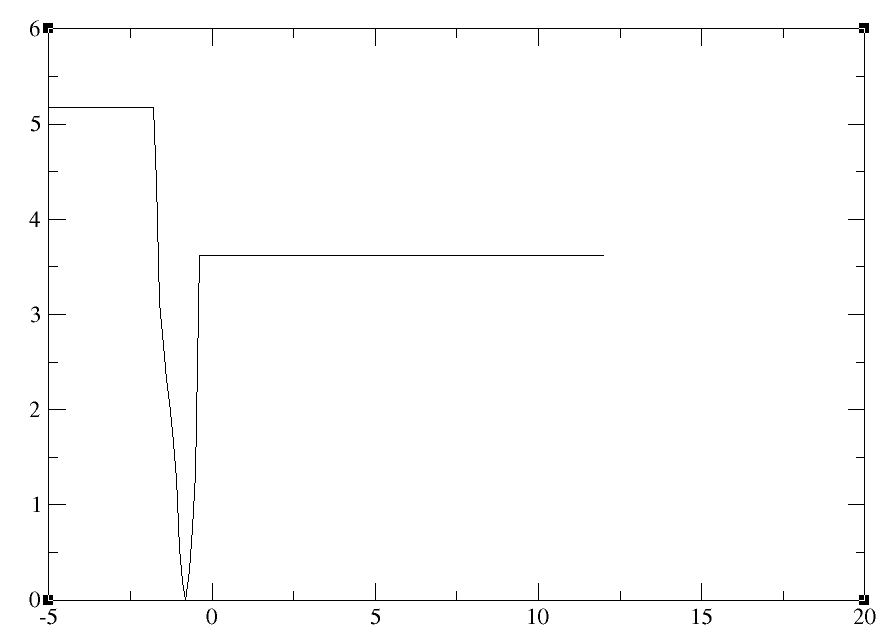
Hello Aashish, the PMF that you show is somewhat expected when you start at
the minimum, and the ABF simulation works its way up. If you were using
metadynamics, you'd get a similar PMF with the only difference that the
baseline would have a the same height at both ends, because the local
metadynamics bias is symmetric (a Gaussian) but the ABF one is not (a local
force).
lowerBoundary and upperBoundary serve two purposes, one to define the range
of the PMF to be binned, and one for applying confining walls. In later
versions of Colvars, these have been split in two distinct features:
https://colvars.github.io/colvars-refman-namd/colvars-refman-namd.html#sec:colvar_grid_params
https://colvars.github.io/colvars-refman-namd/colvars-refman-namd.html#sec:colvarbias_harmonic_walls
and the older syntax that you are using needs to be changed if you switch
to NAMD 2.14 or later:
https://colvars.github.io/colvars-refman-namd/colvars-refman-namd.html#sec:colvars_config_changes
After checking the documentation links above, if there are any specific
questions remaining please bring them up.
One small addendum: the paper that you link cites several papers for ABF
and its implementation, but the paper for the Colvars module itself
actually appeared a few months later:
https://doi.org/10.1080/00268976.2013.813594
Take a look also at other later papers describing or reviewing ABF:
https://doi.org/10.1021/jp506633n
where for example the strategy of using multiple overlapping windows is
described.
On Thu, Oct 8, 2020 at 9:46 AM Aashish Bhatt <aashish.ph16221_at_inst.ac.in>
wrote:
> Dear Sir/ma'am
>
> I have one system Alpha-cyclodextrin with peptide. I want to study
> host-guest interaction.
> for this study, I am following the paper.
> https://pubs.acs.org/doi/pdf/10.1021/jp3128324
>
> Whenever I performing the Adaptive biasing force method I am getting the
> following type of graph for Pmf(Kcal/mol) vs zeta.
>
> [image: image.png]
> I have chosen the heavy atoms of the peptide whose inside the
> alpha-cyclodextrin as the main atoms and second atoms are oxygen
> atoms present in the glycosidic bond.
> I have also query ho to choose lowerboundary and upperboundary.
>
> I have also attached the colvar file.
> Kindly give me some suggestions to understand this issue.
>
> Best Regards
>
> Aashish
>
This archive was generated by hypermail 2.1.6 : Thu Dec 31 2020 - 23:17:14 CST