From: Dhiraj Srivastava (dhirajks_at_gmail.com)
Date: Wed Oct 05 2016 - 14:02:32 CDT
Thank you Everyone.
The simulation was started from crystal structure obtained by
cocrystallizing with ligand. I don't see any significant difference in apo
vs ligand bound form but ligand is an inhibitor. I think for unstable
RMSD, I can blame a small flexible domain. After removing the flexible
domain from RMSD calculation, I got stable RMSD very similar to apo
protein. However its still took long time to equilibrate. the sudden change
in rmsd at around 30 ns is still there. can I use data after 40 ns for the
analysis? is it acceptable to have such a long equilibration time? I am
primarily a crystallographer and I don't have much experience in publishing
articles with MD simulation data. So I don't know what's acceptable in the
community? I am doing simulation to find out if allosteric behaviour of
ligand is due to change in dynamics. I would like to compare it with B
factor change in crystal structure.
Thanks
Dhiraj
On Wed, Oct 5, 2016 at 1:34 PM, Pardis Tabaee <pardis.tabaee.d_at_hotmail.co.uk
> wrote:
> I think it's the average RMSD that was calculated. Maybe you can try to
> equilibrate without the solvent and see if you reach a stable conformation
> of your complex quicker than if you do it with the solvent? There are also
> other strategies. SMD is one of them, where you are applying constraints,
> you can do this with NAMD.
> ------------------------------
> *From:* owner-namd-l_at_ks.uiuc.edu <owner-namd-l_at_ks.uiuc.edu> on behalf of
> Radak, Brian K <bradak_at_anl.gov>
> *Sent:* 05 October 2016 17:18
> *To:* namd-l_at_ks.uiuc.edu; Roshan Shrestha; dhirajks_at_gmail.com
> *Subject:* RE: namd-l: question regarding rmsd
>
> I'm not clear what the "problem" is here. Are you not happy with the
> (large?) oscillations after 100 ns? I don't understand why you would be
> surprised that convergence to a stable state takes that long. I'm more
> surprised that the apo state levels out in 20 ns.
>
> I presume this is RMSD with respect to the initial frame, as is default
> for VMD? That's a completely arbitrary reference and thus I wouldn't hold
> too much stock in it. Is the initial bound state from a crystal structure
> or was it generated by docking to the apo structure? If the latter, then
> there isn't really a reason to believe the initial configuration had a high
> Boltzmann weight anyway - slow equilibration is to be expected and could
> involve large conformational changes.
>
> Brian
>
> Brian Radak
> Postdoctoral Appointee
> Leadership Computing Facility
> Argonne National Laboratory
>
> 9700 South Cass Avenue, Bldg. 240
> Argonne, IL 60439-4854
> (630) 252-8643
> brian.radak_at_anl.gov
> ------------------------------
> *From:* owner-namd-l_at_ks.uiuc.edu [owner-namd-l_at_ks.uiuc.edu] on behalf of
> Roshan Shrestha [roshanpra_at_gmail.com]
> *Sent:* Wednesday, October 05, 2016 11:07 AM
> *To:* namd-l_at_ks.uiuc.edu; dhirajks_at_gmail.com
> *Subject:* Re: namd-l: question regarding rmsd
>
> Don't worry dhiraj, it's obviously rmsd in Y-axis and Time steps in
> X-axis. Shouldn't have been any fuss with it !!!
>
> On Wed, Oct 5, 2016 at 9:48 PM, <dhirajks_at_gmail.com> wrote:
>
>> Sorry. I should have labeled it. Y axis is time in picosecond and x axis
>> is rmsd in Angstrom.
>>
>> Dhiraj
>>
>> Sent from my iPhone
>>
>> On Oct 5, 2016, at 10:55 AM, Pardis Tabaee <pardis.tabaee.d_at_hotmail.co.uk>
>> wrote:
>>
>> Hi,
>>
>>
>> What's on the y axis?
>>
>>
>> Regards,
>>
>>
>> P
>>
>>
>> ------------------------------
>> *From:* owner-namd-l_at_ks.uiuc.edu <owner-namd-l_at_ks.uiuc.edu> on behalf of
>> Dhiraj Srivastava <dhirajks_at_gmail.com>
>> *Sent:* 04 October 2016 22:07
>> *To:* namd-l_at_ks.uiuc.edu
>> *Subject:* namd-l: question regarding rmsd
>>
>> Hi
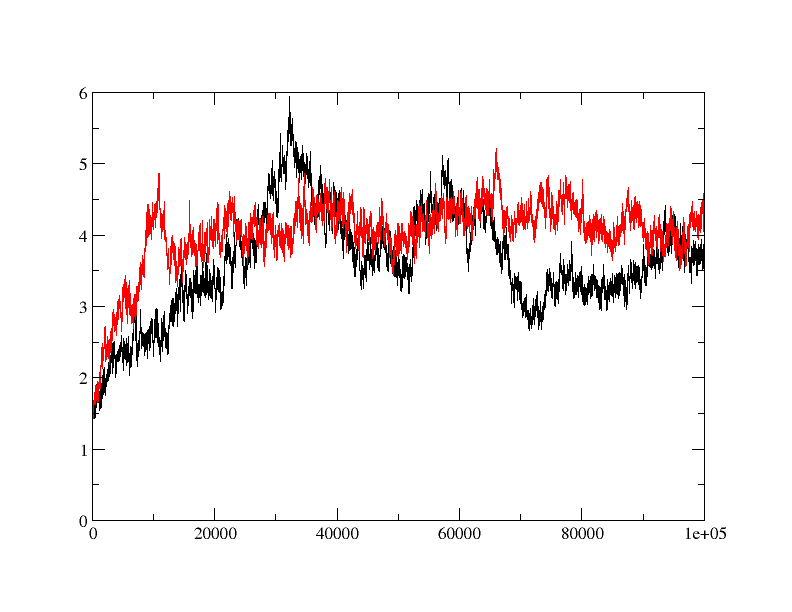
>> I am trying to do MD simulation on a protein with and without ligand.
>> when I did rmsd plot, I found that apo protein is behaving fine (red) but
>> ligand bound form (black) is taking relatively longer time to equilibrate
>> and showing quite a bit of fluctuation in rmsd. is the fluctuation in rmsd
>> value for ligand bound protein is acceptable or is there anything wrong?
>> How can I fix it?
>>
>> thanks
>> Dhiraj
>>
>>
>> [image: Inline image 3]
>>
>>
>
>
> --
> Roshan Shrestha
> Graduate Student
> Central Department of Physics,Tribhuvan University
> Kathmandu,Nepal
>
>
>
This archive was generated by hypermail 2.1.6 : Tue Dec 27 2016 - 23:22:31 CST