From: Martin, Erik W (Erik.Martin_at_stjude.org)
Date: Thu Dec 20 2012 - 11:40:36 CST
I have used this method quite a few times at the start of simulations when I thought it might be required for equilibration. Presently, I was planning on setting up REMD and wanted to get a feel for how my peptide was behaving at different temperatures. I thought a convenient way to do this would be to set up a simulation (starting from a previous restart file) that reassigned the temperatures every 100k steps in the temperature region I plan on using for the replicas.
What NAMD actually ended up doing was somewhat unexpected…
It reassigned the temperatures to my "hold" temp at every defined interval. Then settled to some temperature – the origin of which I can't figure out.
In my configuration file I commented out the Langevin dynamics and included this in the execution:
## EXECUTION SCRIPT
reassignFreq 100000
reassignTemp 270
reassignIncr 10
reassignHold 450
minimize 1000
run 1900000
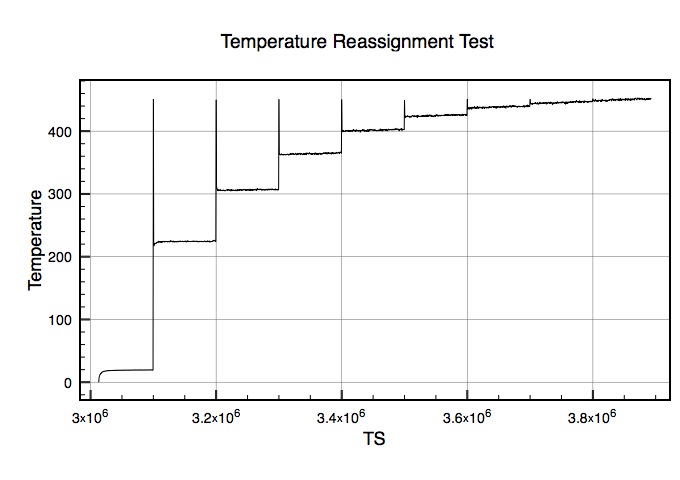
My assumption was that this would start at T=270 and reassign up in increments of 10 degrees every 100k steps until it got to 450. What happened was that every 100k it reassigned to 450 and then settled around the following temperatures:
225>305>360>400>420>435>445>450
Clearly I'm not understanding how this works? I'll attach a plot of the temperature versus time step to help illustrate this problem.
Thanks a lot,
Erik
________________________________
Email Disclaimer: www.stjude.org/emaildisclaimer
Consultation Disclaimer: www.stjude.org/consultationdisclaimer
This archive was generated by hypermail 2.1.6 : Tue Dec 31 2013 - 23:22:52 CST