From: Gianluca Interlandi (gianluca_at_u.washington.edu)
Date: Thu Dec 13 2012 - 16:10:45 CST
Dear James,
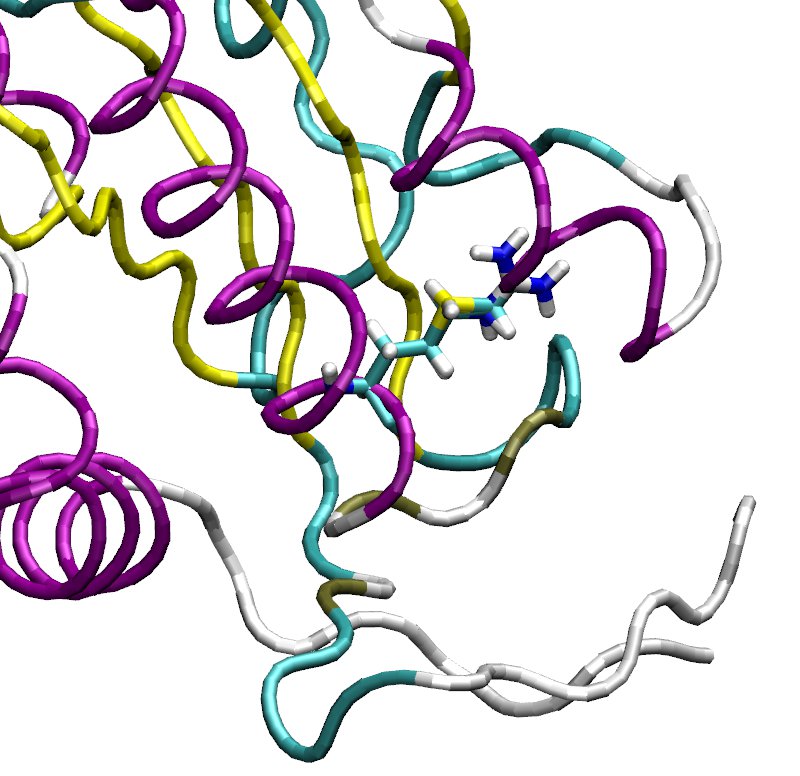
Thanks for your comment. I am not using Alchemify. VMD mutator plugin did
everything for me. The problem I have is that the plugin (when the FEP box
is checked) replaces both the original side chain and the target side
chain in such a way that both (including the original side chain) clash
with the backbone of the protein. I attach a snapshot here to illustrate
this (the mehionine is mutated into arginine). I wished that the mutator
plugin would leave the original side chain in its position (the original
conformation was equilibrated for 10 ns). To solve this problem I ended up
manually creating my own FEP PDB but I still used the PSF created by the
VMD mutator.
Gianluca
On Thu, 13 Dec 2012, JC Gumbart wrote:
> This is irrelevant, because Mutator plus Alchemify make sure that
> the correct exclusion list is established so that the end states of your
> alchemical transformation never see each other.
>
>
> This is outdated - recent versions of NAMD (for at least the last couple years?)
> automatically generates the exclusions without the need to add them to the psf. I
> don't believe there's any need for Alchemify anymore.
> On Dec 13, 2012, at 12:47 PM, Chris Chipot wrote:
>
> Gianluca,
> 1) Is it better to use alchemy in NAMD in the FEP or TI mode
> for this type of calculatons?
>
> Contrary to several extraordinary claims (which have never been
> supported by extraordinary evidences), there is no real advantage
> to use TI over FEP, or the other way around. I would, nonetheless,
> advocate FEP for practical reasons -- you can subsequently use
> parseFEP for your post-analysis, notably get the BAR estimator.
> 2) You describe that in each FEP calculation, 30 intermediate
> states were considered. Does this mean that the progress of
> lambda was broken down into 30 windows each of 150+150 ps?
> How many FEP simulations did you run in total?
>
> Yes to the first question. I do not quite get the second -- the windows
> are chained and the net free-energy change is the sum of free-energy
> changes in individual strata.
> 3) Do I need to use AlchDecouple on or off?
>
> I strongly advocate AlchDecouple off, in particular for solvation
> free-energy calculations, as one simply cannot assume that the
> conformational ensembles in vacuum and in condensed phase
> are identical.
> 4) The VMD mutator plugin replaces both the wild-type and
> mutant side chains such that both clash with the backbone. If
> this is a problem, I might create the PDB myself and still
> use the PSF generated with VMD.
>
> This is irrelevant, because Mutator plus Alchemify make sure that
> the correct exclusion list is established so that the end states of your
> alchemical transformation never see each other.
> Sorry, lots of questions. I would appreciate any help on
> this!
>
> Hopefully, I was convincing enough that it will stop here!
>
>
> Chris Chipot
>
> --
> _______________________________________________________________________
>
> Chris Chipot, Ph.D.
> Theoretical and Computational Biophysics Group
> Beckman Institute
> University of Illinois at Urbana-Champaign
> 405 North Mathews Phone: (217) 244-5711
> Urbana, Illinois 61801 Fax: (217) 244-6078
>
> E-mail: chipot_at_ks.uiuc.edu
> Christophe.Chipot_at_edam.uhp-nancy.fr
> Web: http://www.ks.uiuc.edu/~chipot
> http://www.edam.uhp-nancy.fr
>
> The light shines in the darkness, and the darkness has not overcome it.
> John 1:5.
> _______________________________________________________________________
>
>
>
>
-----------------------------------------------------
Gianluca Interlandi, PhD gianluca_at_u.washington.edu
+1 (206) 685 4435
http://artemide.bioeng.washington.edu/
Research Scientist at the Department of Bioengineering
at the University of Washington, Seattle WA U.S.A.
-----------------------------------------------------
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:22:23 CST