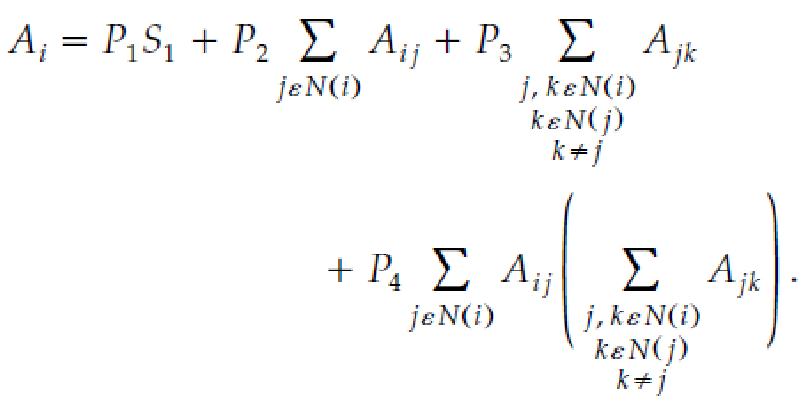
From: Clayton (clj_at_jjwater.info)
Date: Mon Jul 23 2012 - 10:08:35 CDT
NAMD 2.9 had been calculating negative solvent accessible surface areas
for some molecules when when using the Generalized Born Implicit Solvent
algorithm. It is not immediately obvious when this happens because the
SASA is only used internally, as one of the values used in determining
the total electrostatic force (the "ELECT" value reported in the .out
file). I have done some research, picked apart the source code, and
found the problem.
NAMD uses the Linear Combination of Pairwise Overlaps method to
approximate the surface area of molecules. This method models each atom
as a hard sphere, and geometrically calculates the area of each sphere,
then how much of the area of any given sphere is buried inside another.
>From the paper in which this method was first proposed (Jo¨rg Weiser,
Peter S. Shenkin, W. Clark Still, Approximate Atomic Surfaces from
Linear Combinations of Pairwise Overlaps (LCPO), 1998), the formula used
for this is shown in the attached image. A rigid mathematical
derivation seems excessive here, but the meaning of each term is
simple. Ai is the SASA of an individual atom modeled as a hard sphere
i. The first term is simply the area of sphere i. The second term
calculates from the radii and distance between centers of neighbouring
spheres (possibly two spheres of arbitrary size j and k, for this
example), how much surfaces area of sphere i is buried inside its
neighbours, j and k. However, it is possible for spheres j and k to
overlap each other inside i, and in the second term, the area buried
inside both j and k is subtracted twice, which underestimates Ai. The
third term calculates how much area of i is subtracted twice in this
way, and corrects this. Lastly, it is easy to draw three spheres, i, j,
and k, such that j and k both overlap with i, and with each other, but
none of the overlap of j and k is inside i, regardless of how large the
overlap is between j and k. As the distance between i and its two
neighbours decreases, and also as the distance between j and k
decreases, the percentage of the overlap between j and k that is inside
i increases, and fourth term accounts for this. The constants P1
through P4 are constants which are specific to each possible number of
large neighbours for hybridization of each element.
NAMD uses exactly the formula from this paper, and has, at the end of
the source code file "ComputeLCPO.c", a structure which contains all of
the P1 - P4 values for it will need, which are copied exactly from the
appendix of the aforementioned paper. Obviously, for every atom type,
the area of the hard sphere model buried inside neighbors can not exceed
the total area of that sphere. However, the P1 and P2 values for sp3
hybridized N with one large neighbour are such that this is precisely
what NAMD calculates. The P1 value is an order of magnitude smaller
than the P2 value, which makes no physical sense. NAMD subtracts as
"buried" area more area than there is in total for sp3 N with one
neighbour by nearly a factor of 10.
Weiser, Shenkin, and Still calculate these P values three different ways
for each atom, and the numbers they show in the appendix for the very
same atom's P1 value that were arrived at using the other two methods
are entirely sensible. Each is larger than magnitude, but of opposite
sign, from the corresponding P2 value, and an order of magnitude larger
than this value. If one changes the exponent from -2 to -1, this value
will be very close to what was obtained using the other two methods, but
slightly smaller, which is consistent with P1 values obtained using this
method for every other atom in the table. It seems extremely likely
that this is simply a typo in the exponent. However, it may call the
whole table into question. I have tried to contact the authors of the
paper, but haven't heard back.
In any case, the surface area which NAMD calculates for any molecule
containing an sp3 N with one neighbour will be underestimated, and can
be negative if such N atoms make up a large percentage of the area of
the molecule.
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:21:50 CST