The procedures implemented in NAMD are particularly
adapted for performing free
energy calculations that split the ![]() reaction path into a number of non-physical,
intermediate states, or ``windows''. Separate simulations
can be started for each window.
Alternatively, the TCL scripting ability of
NAMD can be employed advantageously
to perform the complete simulation in a single run.
An example, making use of such a script, is supplied at the end
of this section.
However, the setup of sequential alchemical trsnaformations can be
simplified by calling the script library fep.tcl,
found in the lib/alch directory of the NAMD distribution.
This library provides two helper procedures, runFEP to run
a series of evenly spaced windows, and runFEPlist to
specify a list of
reaction path into a number of non-physical,
intermediate states, or ``windows''. Separate simulations
can be started for each window.
Alternatively, the TCL scripting ability of
NAMD can be employed advantageously
to perform the complete simulation in a single run.
An example, making use of such a script, is supplied at the end
of this section.
However, the setup of sequential alchemical trsnaformations can be
simplified by calling the script library fep.tcl,
found in the lib/alch directory of the NAMD distribution.
This library provides two helper procedures, runFEP to run
a series of evenly spaced windows, and runFEPlist to
specify a list of ![]() values to be sampled.
values to be sampled.
The following keywords can be used to run alchemical free energy calculations, whether FEP or TI.
In the current implementation, the electrostatic interactions of an exnihilated, or appearing, particle are linearly coupled to the simulation over the ![]() value range of alchElecLambdaStart - 1.0. At
value range of alchElecLambdaStart - 1.0. At ![]() values less than or equal to the user-defined value of alchElecLambdaStart, electrostatic interactions of the exnihilated particle are fully decoupled from the simulation. Coupling of electrostatic interactions then increases linearly for increasing values of
values less than or equal to the user-defined value of alchElecLambdaStart, electrostatic interactions of the exnihilated particle are fully decoupled from the simulation. Coupling of electrostatic interactions then increases linearly for increasing values of ![]() until
until ![]() =1.0, at which point electrostatic interactions of the exnihilated particle are fully coupled to the simulation.
=1.0, at which point electrostatic interactions of the exnihilated particle are fully coupled to the simulation.
For annihilated, or vanishing, particles the electrostatic interactions are linearly decoupled from the simulation over the ![]() value range of 0 - (1.0 - alchElecLambdaStart). At
value range of 0 - (1.0 - alchElecLambdaStart). At ![]() =0 electrostatic interactions are fully coupled to the simulation, and then linearly decreased with increasing
=0 electrostatic interactions are fully coupled to the simulation, and then linearly decreased with increasing ![]() such that at
such that at ![]() values greater than or equal to (1.0 - alchElecLambdaStart) electrostatic interactions are completely decoupled from the simulation. Two examples, shown in Figure 7, describe the relationship between the user-defined value of
values greater than or equal to (1.0 - alchElecLambdaStart) electrostatic interactions are completely decoupled from the simulation. Two examples, shown in Figure 7, describe the relationship between the user-defined value of ![]() and the coupling of electrostatic or vdW interactions to the simulation.
and the coupling of electrostatic or vdW interactions to the simulation.
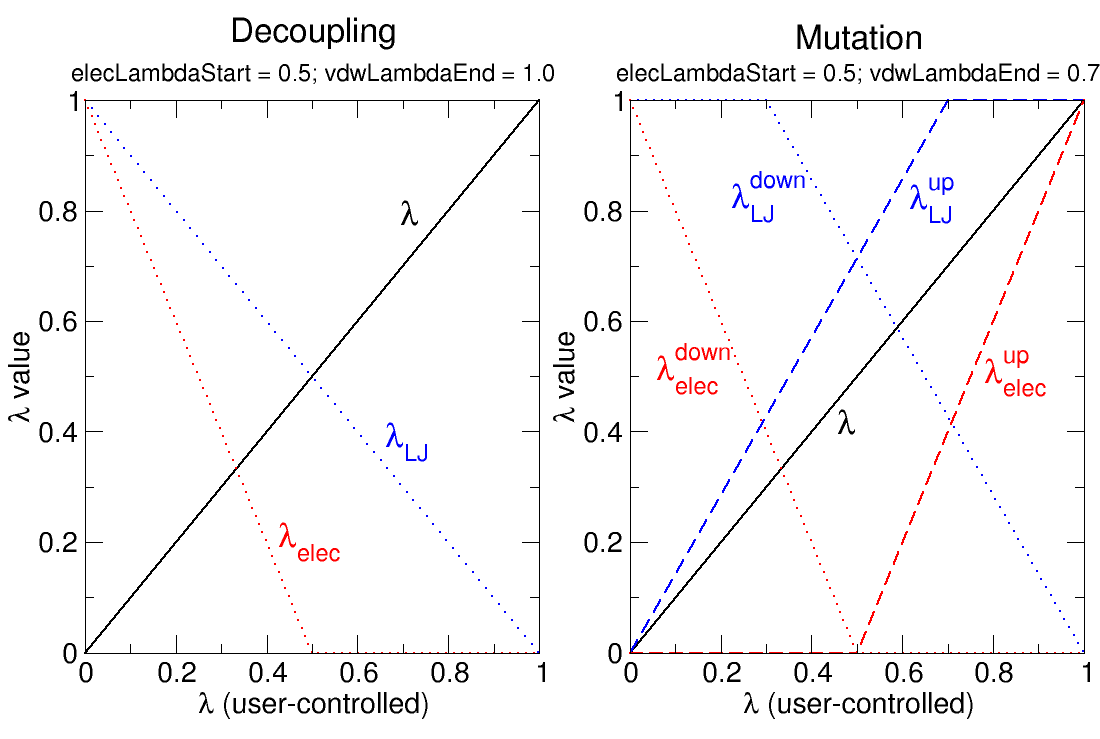 |
For an exnihilated particle, vdW interactions are fully decoupled at ![]() =0. The coupling of vdW interactions to the simulation is then increased with increasing values of
=0. The coupling of vdW interactions to the simulation is then increased with increasing values of ![]() such that at values of
such that at values of ![]() greater than or equal to alchVdwLambdaEnd the vdW interactions of the exnihilated particle are fully coupled to the simulation.
greater than or equal to alchVdwLambdaEnd the vdW interactions of the exnihilated particle are fully coupled to the simulation.
For an annihilated particle, vdW interactions are completely coupled to the simulation for ![]() values between 0 and (1 - alchVdwLambdaEnd). Then, vdW interactions of the annihilated particle are linearly decoupled over the range of
values between 0 and (1 - alchVdwLambdaEnd). Then, vdW interactions of the annihilated particle are linearly decoupled over the range of ![]() values between (1 - alchVdwLambdaEnd) and 1.0. VdW interactions are only fully decoupled when
values between (1 - alchVdwLambdaEnd) and 1.0. VdW interactions are only fully decoupled when ![]() reaches 1.0.
reaches 1.0.
New as of version 2.12: The energy and virial terms added by
LJcorrection on or LJcorrectionAlt on are now also controlled by the vdW ![]() schedule.
The average Lennard-Jones
schedule.
The average Lennard-Jones ![]() and
and ![]() coefficients are computed separately at
both endpoints and then coupled linearly. In most practical situations the
energy difference is extremely negligible, but this is more theoretically sound
than the old behavior of averaging both endpoints together. However, the
kinetic energy component of the virial does still count the endpoints
together, as if annihilated alchemical atoms were an ideal gas. Again, this is
likely quite negligible, nor is it clear that this should be treated specially.
coefficients are computed separately at
both endpoints and then coupled linearly. In most practical situations the
energy difference is extremely negligible, but this is more theoretically sound
than the old behavior of averaging both endpoints together. However, the
kinetic energy component of the virial does still count the endpoints
together, as if annihilated alchemical atoms were an ideal gas. Again, this is
likely quite negligible, nor is it clear that this should be treated specially.