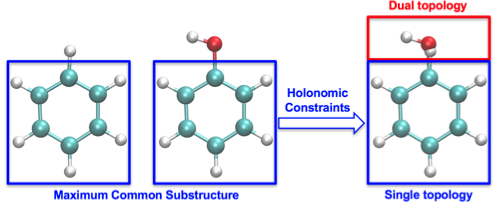
An effective hybrid single-dual topology protocol is designed for the calculation of relative binding affinities of small ligands to a receptor. The protocol was developed as an expansion of the existing dual-topology relative alchemical free energy calculations [50], for either free energy perturbation or thermodynamic integration. In this protocol, the alchemical end states are represented as two separate molecules sharing a common substructure identified through maximum structural mapping. Within the substructure, an atom-to-atom correspondence is established, and each pair of corresponding atoms are holonomically constrained to share identical coordinates at all time throughout the simulation, as shown in Figure 10. The forces are projected and combined at each step for propagation.
|
As it is based on the existing dual topology setup, the major input files
including PDB, PSF and alchemical flag files adopt the same format as
before, with two more partitions accommodating the initial/end states of
the single topology region.
Determining the common substructure generally requires
a special setup tool to determine the maximum structural mapping
that generate the partitions present in the PDB and PSF files.
The dual-topology setup also implements Shobana bonded terms
to support the ring topology change problem [100],
for which a separate input file
lists all unperturbed bonded terms on a ring.
The current implementation supports both relative solvation
free energies of small molecules and relative binding affinities
of drug compounds to proteins.
To enhance sampling of the dual-topology region, the alchemical
calculations can be carried out within a replica-exchange MD scheme
supported by the multiple-copy algorithm module of NAMD, with periodic
attempted swapping of the thermodynamic coupling parameter ![]() betwen neighboring states.
betwen neighboring states.
It needs to be noted that the protocol is currently implemented only on CPU, with a GPU implementation in development. VMD does not yet provide a hybrid topology setup tool, and CHARMM-GUI is testing a beta version (that is not yet available online) to automatically generate all input files for NAMD. For the time being, users can utilize an alternative hybrid structure preparation tool, such as FESetup or AmberTools, and then manually convert the generated CHARMM-formatted input files into a format that can be read by NAMD.
The following keywords enable hybrid single-dual topology simulation.