



Next: Index
Up: NAMD 2.12 User's Guide
Previous: Documentation
Contents
Index
- 1
-
M. P. Allen and D. J. Tildesley.
Computer Simulation of Liquids.
Oxford University Press, New York, 1987.
- 2
-
A. Altis, P. H. Nguyen, R. Hegger, and G. Stock.
Dihedral angle principal component analysis of molecular dynamics
simulations.
J. Chem. Phys., 126(24):244111, 2007.
- 3
-
P. H. Axelsen and D. Li.
Improved convergence in dual-topology free energy calculations
through use of harmonic restraints.
J. Comput. Chem., 19:1278-1283, 1998.
- 4
-
A. Barducci, G. Bussi, and M. Parrinello.
Well-tempered metadynamics: A smoothly converging and tunable
free-energy method.
Phys. Rev. Lett., 100:020603, 2008.
- 5
-
C. H. Bennett.
Efficient estimation of free energy differences with monte carlo
data.
J. Comp. Phys., 22:245-268, 1976.
- 6
-
F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, J. E. F. Meyer, M. D. Brice,
J. R. Rodgers, O. Kennard, T. Shimanouchi, and M. Tasumi.
The protein data bank: A computer-based archival file for
macromolecular structures.
J. Mol. Biol., 112:535-542, 1977.
- 7
-
T. C. Beutler, A. E. Mark, R. C. van Schaik, P. R. Gerber, and W. F. van
Gunsteren.
Avoiding singularities and numerical instabilities in free energy
calculations based on molecular simulations.
Chem. Phys. Lett., 222:529-539, 1994.
- 8
-
D. L. Beveridge and F. M. DiCapua.
Free energy via molecular simulation: Applications to chemical and
biomolecular systems.
Annu. Rev. Biophys. Biophys., 18:431-492, 1989.
- 9
-
A. Bondi.
van der Waals volumes and radii.
J. Phys. Chem., 68:441-451, 1964.
- 10
-
S. Boresch and M. Karplus.
The role of bonded terms in free energy simulations: I. theoretical
analysis.
J. Phys. Chem. A, 103:103-118, 1999.
- 11
-
D. Branduardi, F. L. Gervasio, and M. Parrinello.
From a to b in free energy space.
J Chem Phys, 126(5):054103, 2007.
- 12
-
B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan,
and M. Karplus.
CHARMM: a program for macromolecular energy, minimization, and
dynamics calculations.
J. Comp. Chem., 4(2):187-217, 1983.
- 13
-
A. T. Brünger.
X-PLOR, Version 3.1, A System for X-ray Crystallography and
NMR.
The Howard Hughes Medical Institute and Department of Molecular
Biophysics and Biochemistry, Yale University, 1992.
- 14
-
G. Bussi, A. Laio, and M. Parrinello.
Equilibrium free energies from nonequilibrium metadynamics.
Phys. Rev. Lett., 96(9):090601, 2006.
- 15
-
A. Carter, E, G. Ciccotti, J. T. Hynes, and R. Kapral.
Constrained reaction coordinate dynamics for the simulation of rare
events.
Chem. Phys. Lett., 156:472-477, 1989.
- 16
-
C. Chipot and D. A. Pearlman.
Free energy calculations. the long and winding gilded road.
Mol. Sim., 28:1-12, 2002.
- 17
-
C. Chipot and A. Pohorille, editors.
Free energy calculations. Theory and applications in chemistry
and biology.
Springer Verlag, 2007.
- 18
-
G. Ciccotti, R. Kapral, and E. Vanden-Eijnden.
Blue moon sampling, vectorial reaction coordinates, and unbiased
constrained dynamics.
ChemPhysChem, 6(9):1809-1814, 2005.
- 19
-
J. Comer, J. Phillips, K. Schulten, and C. Chipot.
Multiple-walker strategies for free-energy calculations in namd:
Shared adaptive biasing force and walker selection rules.
J. Chem. Theor. Comput., 10:5276-5285, 2014.
- 20
-
E. A. Coutsias, C. Seok, and K. A. Dill.
Using quaternions to calculate RMSD.
J. Comput. Chem., 25(15):1849-1857, 2004.
- 21
-
E. Darve, D. Rodríguez-Gómez, and A. Pohorille.
Adaptive biasing force method for scalar and vector free energy
calculations.
J. Chem. Phys., 128(14):144120, 2008.
- 22
-
W. K. den Otter.
Thermodynamic integration of the free energy along a reaction
coordinate in cartesian coordinates.
J. Chem. Phys., 112:7283-7292, 2000.
- 23
-
Y. Deng and B. Roux.
Computations of standard binding free energies with molecular
dynamics simulations.
J. Phys. Chem. B, 113(8):2234-2246, 2009.
- 24
-
G. Fiorin, M. L. Klein, and J. Hénin.
Using collective variables to drive molecular dynamics simulations.
Mol. Phys., 111(22-23):3345-3362, 2013.
- 25
-
D. Frenkel and B. Smit.
Understanding Molecular Simulation From Algorithms to
Applications.
Academic Press, California, 2002.
- 26
-
H. Fu, X. Shao, C. Chipot, and W. Cai.
Extended adaptive biasing force algorithm. an on-the-fly
implementation for accurate free-energy calculations.
J. Chem. Theory Comput., 2016.
- 27
-
J. Gao, K. Kuczera, B. Tidor, and M. Karplus.
Hidden thermodynamics of mutant proteins: A molecular dynamics
analysis.
Science, 244:1069-1072, 1989.
- 28
-
M. K. Gilson, J. A. Given, B. L. Bush, and J. A. McCammon.
The statistical-thermodynamic basis for computation of binding
affinities: A critical review.
Biophys. J., 72:1047-1069, 1997.
- 29
-
N. M. Glykos.
Carma: a molecular dynamics analysis program.
J. Comput. Chem., 27(14):1765-1768, 2006.
- 30
-
H. Grubmüller.
Predicting slow structural transitions in macromolecular systems:
Conformational flooding.
Phys. Rev. E, 52(3):2893-2906, Sep 1995.
- 31
-
D. Hamelberg, C. de Oliveira, and J. McCammon.
Sampling of slow diffusive conformational transitions with
accelerated molecular dynamics.
J. Chem. Phys., 127:155102, 2007.
- 32
-
D. Hamelberg, J. Mongan, and J. McCammon.
Accelerated molecular dynamics: a promising and efficient simulation
method for biomolecules.
J. Chem. Phys., 120(24):11919-11929, 2004.
- 33
-
E. Harder, V. M. Anisimov, I. V. Vorobyov, P. E. M. Lopes, S. Y. Noskov, A. D.
MacKerell, and B. Roux.
Atomic level anisotropy in the electrostatic modeling of lone pairs
for a polarizable force field based on the classical drude oscillator.
J. Chem. Theor. Comp., 2(6):1587-1597, 2006.
- 34
-
D. J. Hardy, Z. Wu, J. C. Phillips, J. E. Stone, R. D. Skeel, and K. Schulten.
Multilevel summation method for electrostatic force evaluation.
J. Chem. Theor. Comp., 11:766-779, 2015.
- 35
-
G. D. Hawkins, C. J. Cramer, and D. G. Truhlar.
Parametrized models of aqueous free energies of solvation based on
pairwise descreening of solute atomic charges from a dielectric medium.
J. Phys. Chem., 100:19824-19839, 1996.
- 36
-
J. Hénin and C. Chipot.
Overcoming free energy barriers using unconstrained molecular
dynamics simulations.
J. Chem. Phys., 121:2904-2914, 2004.
- 37
-
J. Hénin, G. Fiorin, C. Chipot, and M. L. Klein.
Exploring multidimensional free energy landscapes using
time-dependent biases on collective variables.
J. Chem. Theory Comput., 6(1):35-47, 2010.
- 38
-
T. Huber, A. E. Torda, and W. van Gunsteren.
Local elevation - A method for improving the searching properties
of molecular-dynamics simulation.
Journal of Computer-Aided Molecular Design, 8(6):695-708, DEC
1994.
- 39
-
M. Iannuzzi, A. Laio, and M. Parrinello.
Efficient exploration of reactive potential energy surfaces using
car-parrinello molecular dynamics.
Phys. Rev. Lett., 90(23):238302, 2003.
- 40
-
W. Jiang, D. Hardy, J. Phillips, A. MacKerell, K. Schulten, and B. Roux.
High-performance scalable molecular dynamics simulations of a
polarizable force field based on classical Drude oscillators in NAMD.
J. Phys. Chem. Lett., 2:87-92, 2011.
- 41
-
J. Kastner and W. Thiel.
Bridging the gap between thermodynamic integration and umbrella
sampling provides a novel analysis method: ``umbrella integration''.
J. Chem. Phys., 123(14):144104, 2005.
- 42
-
P. M. King.
Free energy via molecular simulation: A primer.
In W. F. Van Gunsteren, P. K. Weiner, and A. J. Wilkinson, editors,
Computer simulation of biomolecular systems: Theoretical and
experimental applications, volume 2, pages 267-314. ESCOM, Leiden, 1993.
- 43
-
J. G. Kirkwood.
Statistical mechanics of fluid mixtures.
J. Chem. Phys., 3:300-313, 1935.
- 44
-
P. A. Kollman.
Free energy calculations: Applications to chemical and biochemical
phenomena.
Chem. Rev., 93:2395-2417, 1993.
- 45
-
E. A. Koopman and C. P. Lowe.
Advantages of a Lowe-Andersen thermostat in molecular dynamics
simulations.
J. Chem. Phys., 124:204103, 2006.
- 46
-
A. Laio and M. Parrinello.
Escaping free-energy minima.
Proc. Natl. Acad. Sci. USA, 99(20):12562-12566, 2002.
- 47
-
G. Lamoureux, E. Harder, I. V. Vorobyov, B. Roux, and A. D. MacKerell.
A polarizable model of water for molecular dynamics simulations of
biomolecules.
Chem. Phys. Lett., 418(1-3):245-249, 2006.
- 48
-
G. Lamoureux and B. Roux.
Modeling induced polarization with classical Drude oscillators:
Theory and molecular dynamics simulation algorithm.
J. Chem. Phys., 119(6):3025-3039, 2003.
- 49
-
A. Lesage, T. Lelièvre, G. Stoltz, and J. Hénin.
Smoothed biasing forces yield unbiased free energies with the
extended-system adaptive biasing force method.
J. Phys. Chem. B.
(submitted).
- 50
-
N. Lu, D. A. Kofke, and T. B. Woolf.
Improving the efficiency and reliability of free energy perturbation
calculations using overlap sampling methods.
J. Comput. Chem., 25:28-39, 2004.
- 51
-
Z. M., T. P. Straatsma, and M. J. A.
Separation-shifted scaling, a new scaling method for
Lennard-Jones interactions in thermodynamic integration.
J. Chem. Phys., 100:9025-9031, 1994.
- 52
-
A. E. Mark.
Free energy perturbation calculations.
In P. v. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A.
Kollman, H. F. Schaefer III, and P. R. Schreiner, editors, Encyclopedia
of computational chemistry, volume 2, pages 1070-1083. Wiley and Sons,
Chichester, 1998.
- 53
-
S. J. Marrink, A. H. de Vries, and A. E. Mark.
Coarse grained model for semiquantitative lipid simulations.
J. Phys. Chem. B, 108:750-760, 2004.
- 54
-
S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman, and A. H. de Vries.
The martini forcefield: coarse grained model for biomolecular
simulations.
J. Phys. Chem. B, 111:7812-7824, 2007.
- 55
-
J. A. McCammon and S. C. Harvey.
Dynamics of Proteins and Nucleic Acids.
Cambridge University Press, Cambridge, 1987.
- 56
-
K. Minoukadeh, C. Chipot, and T. Lelièvre.
Potential of mean force calculations: A multiple-walker adaptive
biasing force approach.
J. Chem. Theor. Comput., 6:1008-1017, 2010.
- 57
-
L. Monticelli, S. Kandasamy, X. Periole, and R. L. D. T. S. Marrink.
The martini coarse grained forcefield: extension to proteins.
J. Chem. Theor. Comp., 4:819-834, 2008.
- 58
-
Y. Mu, P. H. Nguyen, and G. Stock.
Energy landscape of a small peptide revealed by dihedral angle
principal component analysis.
Proteins, 58(1):45-52, 2005.
- 59
-
J. K. Noel, P. C. Whitford, K. Y. Sanbonmatsu, and J. N. Onuchic.
SMOG@ctbp: simplified deployment of structure-based models in
GROMACS.
Nucleic Acids Research, 38:W657-61, 2010.
- 60
-
A. Onufriev, D. Bashford, and D. A. Case.
Modification of the generalised born model suitable for
macromolecules.
J. Phys. Chem., 104:3712-3720, 2000.
- 61
-
A. Onufriev, D. Bashford, and D. A. Case.
Exploring protein native states and large-scale conformational
changes with a modified generalized born model.
Proteins: Struct., Func., Gen., 55:383-394, 2004.
- 62
-
D. A. Pearlman.
A comparison of alternative approaches to free energy calculations.
J. Phys. Chem., 98:1487-1493, 1994.
- 63
-
J. W. Pitera and J. D. Chodera.
On the use of experimental observations to bias simulated ensembles.
J. Chem. Theory Comput., 8:3445-3451, 2012.
- 64
-
P. Raiteri, A. Laio, F. L. Gervasio, C. Micheletti, and M. Parrinello.
Efficient reconstruction of complex free energy landscapes by
multiple walkers metadynamics.
J. Phys. Chem. B, 110(8):3533-9, 2005.
- 65
-
A. Roitberg and R. Elber.
Modeling side chains in peptides and proteins: Application of the
locally enhanced sampling technique and the simulated annealing methods to
find minimum energy conformations.
J. Chem. Phys., 95:9277-9287, 1991.
- 66
-
M. J. Ruiz-Montero, D. Frenkel, and J. J. Brey.
Efficient schemes to compute diffusive barrier crossing rates.
Mol. Phys., 90:925-941, 1997.
- 67
-
M. Schaefer and C. Froemmel.
A precise analytical method for calculating the electrostatic energy
of macromolecules in aqueous solution.
J. Mol. Biol., 216:1045-1066, 1990.
- 68
-
R. Shen, W. Han, G. Fiorin, S. M. Islam, K. Schulten, and B. Roux.
Structural refinement of proteins by restrained molecular dynamics
simulations with non-interacting molecular fragments.
PLoS Comput. Biol., 11(10):e1004368, 2015.
- 69
-
M. R. Shirts, D. L. Mobley, J. D. Chodera, and V. S. Pande.
Accurate and efficient corrections for missing dispersion
interactions in molecular simulations.
J. Phys. Chem. B, 111(45):13052-13063, 2007.
- 70
-
C. Simmerling, T. Fox, and P. A. Kollman.
Use of locally enhanced sampling in free energy calculations:
Testing and application to the
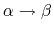 anomerization of
glucose.
anomerization of
glucose.
J. Am. Chem. Soc., 120(23):5771-5782, 1998.
- 71
-
C. Simmerling, M. R. Lee, A. R. Ortiz, A. Kolinski, J. Skolnick, and P. A.
Kollman.
Combining MONSSTER and LES/PME to predict protein structure from
amino acid sequence: Application to the small protein CMTI-1.
J. Am. Chem. Soc., 122(35):8392-8402, 2000.
- 72
-
R. D. Skeel and J. J. Biesiadecki.
Symplectic integration with variable stepsize.
Ann. Numer. Math., 1:191-198, 1994.
- 73
-
J. Srinivasan, M. W. Trevathan, P. Beroza, and D. A. Case.
Application of a pairwise generalized born model to proteins and
nucleic acids: inclusion of salt effects.
Theor Chem Acc, 101:426-434, 1999.
- 74
-
W. C. Still, A. Tempczyk, R. C. Hawley, and T. Hendrickson.
Semianalytical treatment of solvation for molecular mechanics and
dynamics.
J. Am. Chem. Soc., 112:6127-6129, 1990.
- 75
-
T. P. Straatsma and J. A. McCammon.
Multiconfiguration thermodynamic integration.
J. Chem. Phys., 95:1175-1118, 1991.
- 76
-
T. P. Straatsma and J. A. McCammon.
Computational alchemy.
Annu. Rev. Phys. Chem., 43:407-435, 1992.
- 77
-
B. T. Thole.
Molecular polarizabilities calculated with a modified dipole
interaction.
Chem. Phys., 59:341-350, 1981.
- 78
-
P. Van Duijnen and M. Swart.
Molecular and atomic polarizabilities: Thole's model revisited.
J. Phys. Chem. A, 102(14):2399-2407, 1998.
- 79
-
W. F. van Gunsteren.
Methods for calculation of free energies and binding constants:
Successes and problems.
In W. F. Van Gunsteren and P. K. Weiner, editors, Computer
simulation of biomolecular systems: Theoretical and experimental
applications, pages 27-59. Escom, The Netherlands, 1989.
- 80
-
Y. Wang, C. Harrison, K. Schulten, and J. McCammon.
Implementation of accelerated molecular dynamics in NAMD.
"Comp. Sci. Discov.", 4:015002, 2011.
- 81
-
J. Weiser, P. Senkin, and W. C. Still.
Approximate atomic surfaces from linear combinations of pairwise
overlaps (LCPO).
J. Comp. Chem., 20:217-230, 1999.
- 82
-
A. D. White and G. A. Voth.
Efficient and minimal method to bias molecular simulations with
experimental data.
J. Chem. Theory Comput., ASAP, 2014.
- 83
-
P. C. Whitford, J. K. Noel, S. Gosavi, A. Schug, K. Y. Sanbonmatsu, and J. N.
Onuchic.
An all-atom structure-based potential for proteins: Bridging minimal
models with all-atom empirical forcefields.
Proteins, 75(2):430-441, 2009.
- 84
-
P. C. Whitford, A. Schug, J. Saunders, S. P. Hennelly, J. N. Onuchic, and K. Y.
Sanbonmatsu.
Nonlocal helix formation is key to understanding
s-adenosylmethionine-1 riboswitch function.
Biophysical Journal, 96(2):L7 - L9, 2009.
- 85
-
L. Zheng and W. Yang.
Practically efficient and robust free energy calculations:
Double-integration orthogonal space tempering.
J. Chem. Theor. Compt., 8:810-823, 2012.
- 86
-
R. W. Zwanzig.
High-temperature equation of state by a perturbation method. i.
nonpolar gases.
J. Chem. Phys., 22:1420-1426, 1954.
http://www.ks.uiuc.edu/Research/namd/